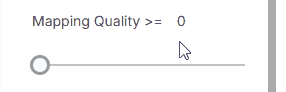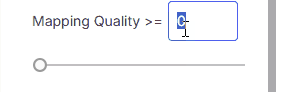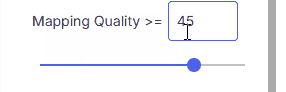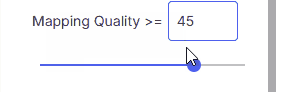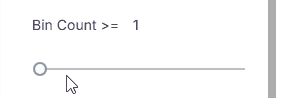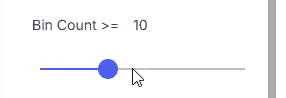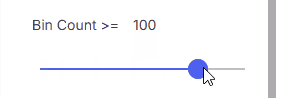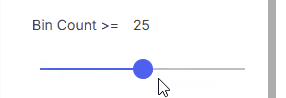# Quality filters
Use Quality filters to refine variant reviews by setting **quality thresholds** and filtering based on **calling methodology**. This prioritizes variants that meet specific confidence metrics for interpretation.
The Quality filters can operate in a [Simple ](#simple-mode)or [Advanced ](#advanced-mode)mode.
{% hint style="warning" %}
**Simple and advanced filtering modes operate independently.**
Even with similar settings, results may differ due to variations in filtering logic and options available.
{% endhint %}
{% hint style="success" %}
**Tips:**
* Start with **Simple Mode** for quick triaging, then refine using **Advanced Mode**
* Always cross-check low-quality calls in **IGV visualization** before discarding
{% endhint %}
## Simple mode
{% columns %}
{% column width="41.66666666666667%" valign="middle" %}
{% endcolumn %}
{% column width="58.33333333333333%" %}
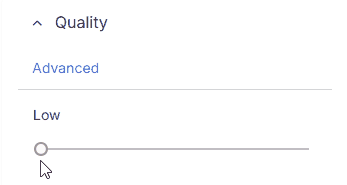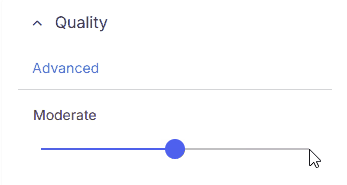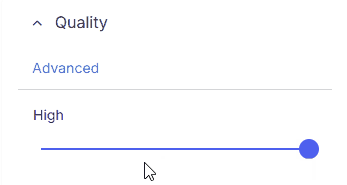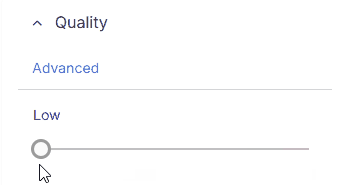
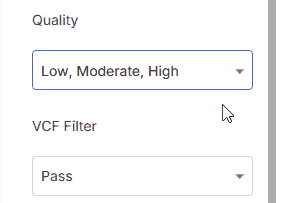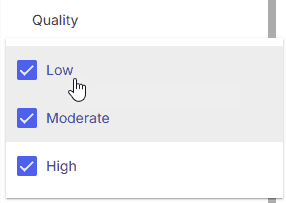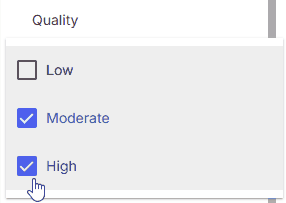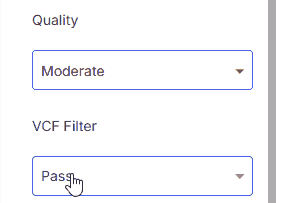
Quickly filter variants by minimum overall sequencing quality:
* **Low**: No quality filtering applied
* **Moderate**: Includes moderate and high quality variants with read depth ≥10
* **High**: Includes only high quality variants with read depth ≥10
{% endcolumn %}
{% endcolumns %}
## Advanced mode
Provides fine-grained control over multiple quality metrics depending on the variant type.
### Quality
{% columns %}
{% column width="41.66666666666667%" valign="middle" %}
{% endcolumn %}
{% column width="58.33333333333333%" valign="middle" %}
Aggregated quality specific to variant type and caller. Determines overall confidence score thresholds for all variant types.
* **Low**
* **Moderate**
* **High**
{% endcolumn %}
{% endcolumns %}
### VCF FILTER
{% columns %}
{% column width="41.66666666666667%" valign="middle" %}
{% endcolumn %}
{% column width="58.33333333333333%" valign="middle" %}
FILTER column value in VCF.
* **Pass**: Variants with PASS value
* **Other values**: Variants with any value except PASS
{% endcolumn %}
{% endcolumns %}
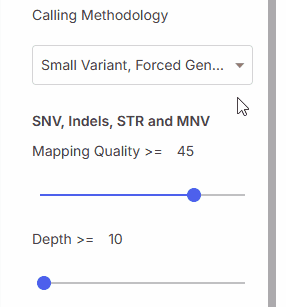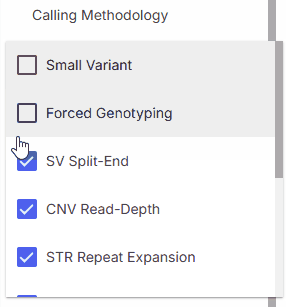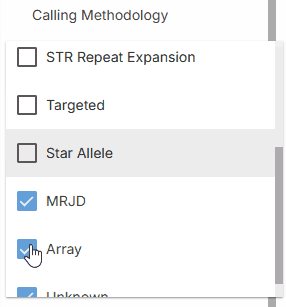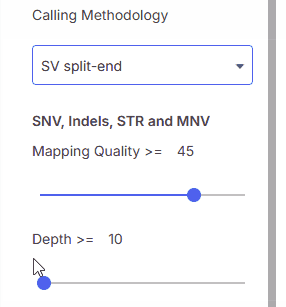
### Calling methodology
{% columns %}
{% column width="41.66666666666667%" valign="middle" %}
{% endcolumn %}
{% column width="58.33333333333333%" valign="middle" %}
Calling methodology filter allows filtering by [variant caller](https://help.emg.illumina.com/emedgene-analyze-manual/supported-vcfs-variant-callers) type.
* **Small Variant**
* **CNV Read-Depth**
* **Forced Genotyping**
* **Star Allele**
* **STR Repeat Expansion**
* **SV Split-End**
* **Targeted**
* **MRJD**
* **Array**
* **Unknown**
{% hint style="success" %}
**Note:** This is especially useful for filtering CNVs from different callers (e.g., DRAGEN Read-Depth vs. SV Caller) to apply separate noise databases or QC settings.
{% endhint %}
{% endcolumn %}
{% endcolumns %}
### Quality metrics by variant type: **SNV, indel, MNV (v100.39.0+), and STR**
#### Mapping quality (MQ)
{% columns %}
{% column width="41.66666666666667%" valign="middle" %}
{% endcolumn %}
{% column width="58.33333333333333%" valign="middle" %}
Minimum mapping quality.
* **Range**: 0–60 using the slider
* **Custom value**: Enter a number in the input field (value can exceed 60)
{% endcolumn %}
{% endcolumns %}
#### Depth (DP)
{% columns %}
{% column width="41.66666666666667%" valign="middle" %}
{% endcolumn %}
{% column width="58.33333333333333%" valign="middle" %}
Minimum read depth.
* **Range**: 0–500 using the slider
* **Custom value**: Enter a number in the input field (value can exceed 500)
{% endcolumn %}
{% endcolumns %}
#### Alternate read count
{% columns %}
{% column width="41.66666666666667%" valign="middle" %}
{% endcolumn %}
{% column width="58.33333333333333%" valign="middle" %}
Minimum number of reads supporting the alternate allele.
* **Range**: 0–500 using the slider
* **Custom value**: Enter a number in the input field (value can exceed 500)
{% endcolumn %}
{% endcolumns %}
#### Allele bias
{% columns %}
{% column width="41.66666666666667%" valign="middle" %}
{% endcolumn %}
{% column width="58.33333333333333%" valign="middle" %}
Minimum and maximum allowable allele bias. For mtDNA, this represents heteroplasmy level.
* **Range:** 0–100 using two sliders (minimum and maximum)
{% hint style="warning" %}
When analyzing a VCF file that includes **CNV variants with** [**AD**](#user-content-fn-1)[^1] **values**, be aware that these variants—along with SNVs—may also be impacted by the allele bias filter.
{% endhint %}
{% endcolumn %}
{% endcolumns %}
### Quality metrics by variant type: **SV, CNV, and LOH/ROH**
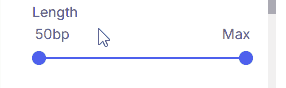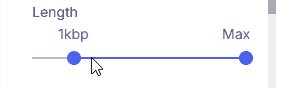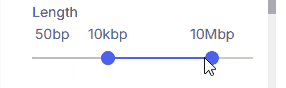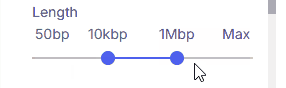
#### Length
{% columns %}
{% column width="41.66666666666667%" valign="middle" %}
{% endcolumn %}
{% column width="58.33333333333333%" %}
Sets the minimum and maximum variant size using two sliders.
* **Supported values**: 50 bp, 1 kbp, 10 kbp, 100 kbp, 1 Mbp, 10 Mbp, Max.
* **Max:** When selected as the upper threshold, all CNVs 50 bp and larger are shown.
{% endcolumn %}
{% endcolumns %}
#### Bin Count
{% columns %}
{% column width="41.66666666666667%" valign="middle" %}
{% endcolumn %}
{% column width="58.33333333333333%" %}
Minimum bin count.
* **Range**: 0–500 using the slider
* **Custom value**: Enter a number in the input field (value can exceed 500)
{% endcolumn %}
{% endcolumns %}
{% hint style="warning" %}
When analyzing a VCF file that includes CNV variants with AD[^1] values, be aware that these variants—along with SNVs—may also be impacted by the **Allele bias** **filter**.
{% endhint %}
## Default values
When filters are reset to default, the Quality filters are set to:
Filter
Setting
Quality
Moderate, High
Mapping Quality
≥45
Depth
≥10
Other Quality filters
No restriction
[^1]: Allele Depth — total read depth for each allele according to the Variant Call Format specification.