# New in Emedgene V100.39.0 (October 16th, 2025)
### Introduction
These Release Notes detail the key new features, enhancements, and bug fixes available in Emedgene v100.39.0.
Note on version naming: Emedgene has now brought back major versioning and releases will follow major.minor.patch conventions. This is as part of preparations for an IVDR submission in the EU. There are no changes to our release policy, both major and minor versions are selectable by customers.
Release highlights:
* Support for DRAGEN 4.4 from VCF. DRAGEN 4.4 boosts CNV capabilities with an Allele Specific Copy Number CNV caller, provides a 30% improvement to SV calling accuracy, has new in-run PON generation capabilities and new targeted callers for WES.
* Updates to the automated ACMG classification module for SNVs for PS1, BS2, PP1/BS4,BP5, and the possibility to exclude PP5/BP6 from the auto-calculation. All tags include improved evidence.
* This module is now annotated with the gene-specific ClinGen VCEP guidelines, and an alert appears on variants that require custom curation.
* Major improvements to the Emedgene variant and gene external links including Google Scholar, Pubmed, Litvar for publication searches and GeneCC and Monarch for diseases. Importantly, GeneCC includes gene-disease validity strength.
* Improved support for MNVs. Emedgene now supports MNV ingestion, annotation and filtering, and each variant is displayed as an MNV and is also split and annotated individually.
* Our data sharing capabilities now allow cross-cloud networking, for labs with collaborators outside their cloud region. This is an opt-in, lab-controlled capability, where sharing permissions can be defined granularly per network.
* CNV/cytogenetic interpretation improvements including a combination analysis tools and IGV visualization component, support for DRAGEN Array 1.3 including mosaicism and expanded chromosome quality metrics.
* Additional voice-of-customer improvements:
* Improvements to the transcript prioritization logic to support customers running panels.
* Genome pipeline speed has been reduced by an additional 30%, so that a typical genome will run in 2:15 hrs.
* Secondary findings updated to v3.3.
* ClinVar updates will now only retain submissions contributing to the aggregate classification.
* Emedgene default Full Genes and Clinical Regions BED files have been updated.
* Self-serve: Users can delete Curate variants, have an improved preset management interface, and can self-configure BYOK integrations.
Emedgene customers can select their preferred version out of any of the past 5 releases. Customers on v34.0 and below should select an upgrade path at this time. V34 end-of-life is October 31st.
The software release includes the following components, which can be selected independently:
* Workbench 100.39
* Pipeline 100.39
Patches
Date
V100.39.300
February 26, 2026
V100.39.2
December 28, 2025
V100.39.1
November 12, 2025
## Support for DRAGEN 4.4 from VCF
New Features:
This version supports [DRAGEN 4.4](https://help.dragen.illumina.com/product-guide/dragen-v4.4) for customers starting from VCF. DRAGEN 4.4 improvements include:
* Small Variants: Personalized pangenome and ML model result in a 17.76% reduction in FP+FN.
* New! Allele Specific Copy Number (ASCN) caller improves accuracy of CNV calling and outputs LOH segments which are ingested as reportable events in Emedgene.
* New! Cytogenetics Modality when enabled on the ASCN CNV caller on WGS achieves CMA-equivalent results.
* New! In-run PON improves sensitivity and specificity of PON, and we’ve also added PON correlation metrics to the DRAGEN report.
* New! SMN & HBA targeted callers for spiked Illumina Exome 2.5.
* 30% boost to SV calling accuracy and Emedgene now displays the Imprecise tag.
New in Emedgene:
* Support for the Ploidy caller, from DRAGEN 4.4, 4.3 and 4.2 (when starting from VCF).
* Support for HLA genes from the Star Allele caller.
* Many new quality metrics exposed in the Emedgene user interface.
### General compatibility with DRAGEN 4.4 outputs
The following table summarizes the supported DRAGEN 4.4 callers:
| Type | DRAGEN Output | FASTQ | VCF (BYOD) | Notes |
| ------------------------------------------------------------------- | --------------------------------------- | ----- | ---------- | ------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------- |
| BAM/CRAM |
In-.BAM/.CRAM
Out - .CRAM
| ✕ | ✓ | Requires .bai in same folder |
| Small variants | In-vcf/gvcf Out-hard-filtered.gvcf.gz | ✕ | ✓ | *Targeted caller variants are removed and ingested via the targeted vcf.* |
| SV del/dup/ins | sv.vcf.gz | ✕ | ✓ | *VNTR caller outputs are removed from the SV output and not supported on Emedgene yet.* |
|
CNV
CNV ASCN
CNV ASCN with Cytogenetics Modality
| cnv.vcf.gz | ✕ | ✓\* |
When running the new CNV ASCN command line, Emedgene will ingest del, dup and LOH variants.
\*The new CNV-SV merged file is also supported. Do not use both the CNV and CNV-SV file.
|
|
CNV-SV
CNV-SV ASCN
| cnv\_sv.vcf.gz | ✕ | ✓\* |
When running the new CNV ASCN command line, Emedgene will ingest del, dup and LOH variants.
\*The new CNV-SV merged file is also supported. Do not use both the CNV and CNV-SV file.
|
| STR | repeats.vcf.gz | ✕ | ✓ |
Do not use the ExpansionHunter SMN caller, this will fail the case.
| ✕ | ✓ | SMN, GBA, HBA, CYP21A2 w/o CNV, supported. Need to push both files to get SNV and CNV data. |
| Star Allele | Targeted.json | ✕ | ✓ | Star allele caller, CYP2D6 & CYP2B6 and HLA genes are supported. |
| Ploidy | ploidy.vcf | ✕ | ✓ | |
| Ploidy | ploidy\_estimation\_metrics.csv | ✕ | ✕ | Security requirements prevent the ingestion of csv files at this time, can be pushed in tar. |
| QC metrics | mapping\_metrics.csv | ✕ | ✕ | Security requirements prevent the ingestion of csv files at this time, can be pushed in tar. |
| QC metrics | bed\_coverage\_metrics.csv | ✕ | ✕ | Metrics file containing FASTQC information. |
| QC metrics/TAR | \*.metrics.tar.gz | ✕ | ✓\* |
DRAGEN report for customers starting from VCF. Only available via API. Tar file must contain one of the following.
METRICS\_PATTERNS = \[
r'.csv$',
r'.tsv$',
r'.counts(.gc-corrected)?(.gz)?$',
r'.(ploidy
|
| ROH Viz | roh.bed | ✕ | ✓ | |
| BAF BigWig | hard-filtered.baf.bw | ✕ | ✓ | B-Allele frequency (BAF) output. |
| TNS BigWig | tn.bw | ✕ | ✓ | Bigwig representation of the tangent normalized signal. |
| Target Counts BigWig | target.counts.bw | ✕ | ✓ | BigWig representation of the target counts bins. |
### Supported new CNV callers in DRAGEN 4.4
DRAGEN 4.4 has added many new CNV calling improvements.
The **CNV Allele Specific Copy Number (ASCN) caller** is available for WGS only, and leverages depth of coverage and B-allele frequencies (BAF) to detect germline copy number aberrations and regions with absence of heterozygosity (AOH). Full [DRAGEN documentation](https://help.dragen.illumina.com/product-guide/dragen-v4.4/dragen-dna-pipeline/cnv-calling/available-pipelines/germline-cnv-calling-wgs-ascn) is available including limitations.
All of the caller outputs, deletions, duplications and LOH, are supported in Emedgene.
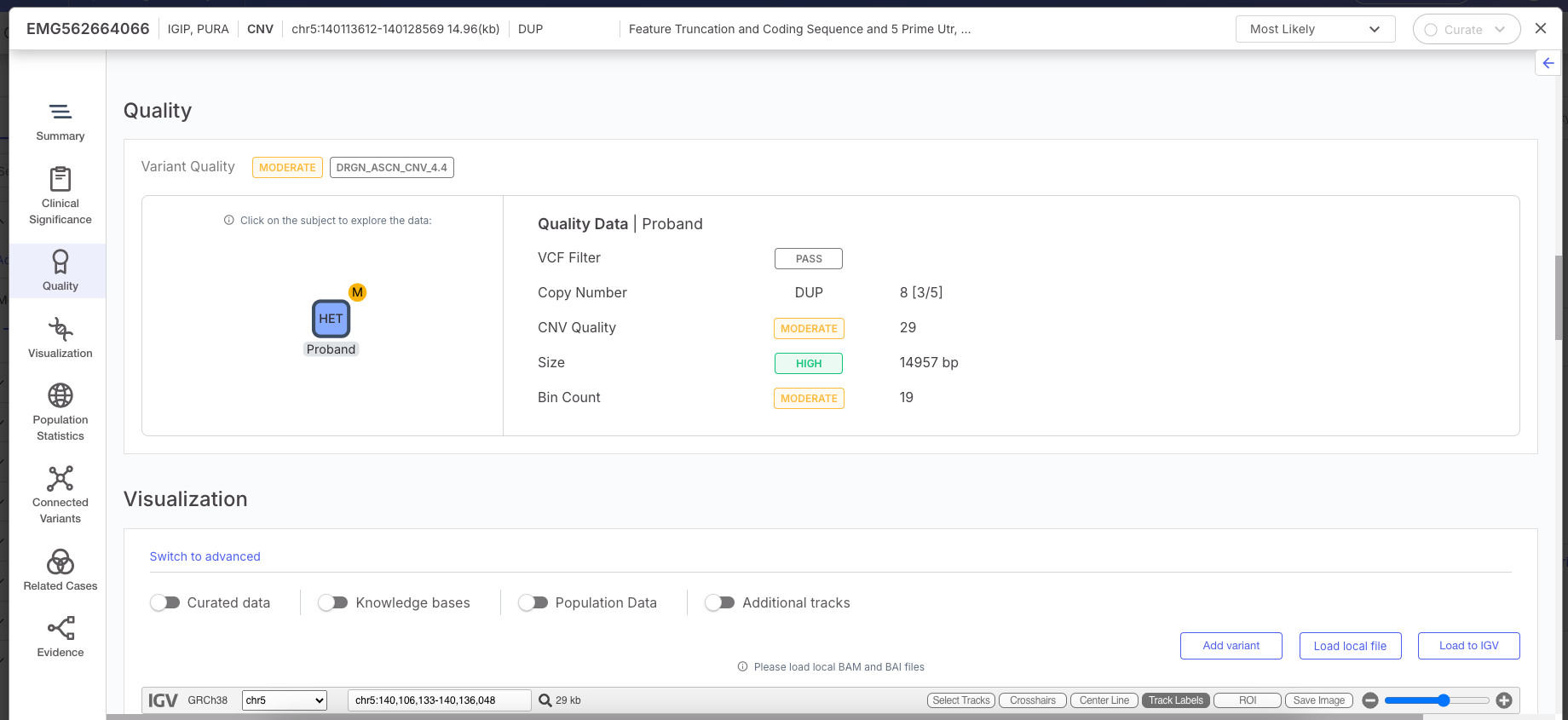
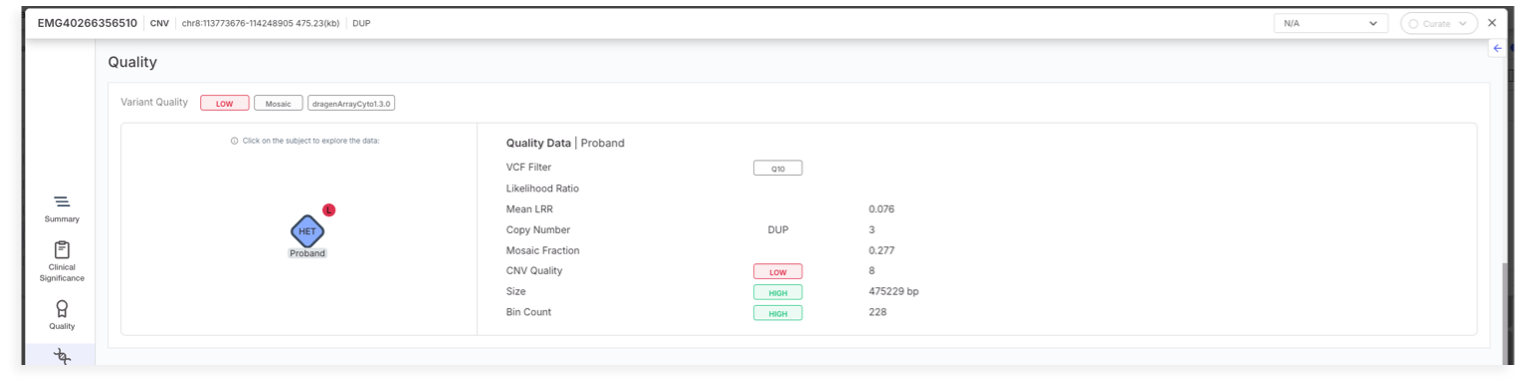
The caller outputs a Minor Allele Copy Number estimation which is displayed in the Emedgene Variant Page | Quality tab, in the Copy Number field, and aims to provide more transparent and informative quality metrics for each variant.
The new display will show a format like \[X/Y], where:
* X represents the *Minor Copy Number (MCN)* retrieved from the VCF.
* Y is calculated by subtracting MCN from the total *Copy Number (CN)* (for example, if CN = 4 and MCN = 1, the display will show \[1/3]).
* If no MCN is available for a particular variant, this additional information will simply not be shown.
This caller also provides Mosaic Alterations Detection, and those variants will display the Mosaic tag in Emedgene, which is also available for filtering.
This caller follows the [VCFv4.4 specs](https://samtools.github.io/hts-specs/VCFv4.4.pdf) which are now fully supported in Emedgene.
Customers can also run the CNV ASCN command line as part of the CNV-SV caller, and this is supported in Emedgene.
{% hint style="info" %}
Limitation: Emedgene does not support the CNV ASCN caller when customer downgrade to the VCF v4.2 spec.
{% endhint %}
The CNV ASCN caller is also available in a **Cytogenetics Modality**. The [Cytogenetics modality for the CNV caller](https://help.dragen.illumina.com/product-guide/dragen-v4.4/dragen-dna-pipeline/cnv-calling/additional-documentation/cytogenetics-modality) allows the user to visualize CNAs at different resolutions, aiming at providing a more flexible workspace for different use cases.
The new Cytogenetics Modality caller has been evaluated as a replacement for CMA as a first-tier test. [Preprint](https://doi.org/10.1101/2025.05.24.25328260) with Broad and Quest available, key findings:
* High Concordance with CMA: WGS achieved 97.28% concordance with CMA for clinically relevant CNVs and LOH.
* WGS provided better breakpoint resolution.
* WGS covered over 97% of clinically relevant regions for CNV detection, compared to less than 3% with CMA.
All of the CNV ASCN caller features are available for the Cytogenetics Modality caller as well.
{% hint style="info" %}
Limitation: All events with the SEGID tag (whole chromosome arm) will be designated as low quality in Emedgene.
{% endhint %}
### In-run PON workflow and PON correlation metrics
DRAGEN v4.4 adds workflow-level support for automatically constructing a PoN (Panel of Normals) from a batch of samples on ICA/BSSH. This new workflow provides batch specific artefact correction and reduces putative false positives by as much as 65% compared to WGS CNV while maintaining recall. It also eliminates need for pre-curated/pre-built normal panel.
Emedgene supports ingestion of VCFs generated from these workflows.
#### Panel-of-Normals Correlation in DRAGEN Reports
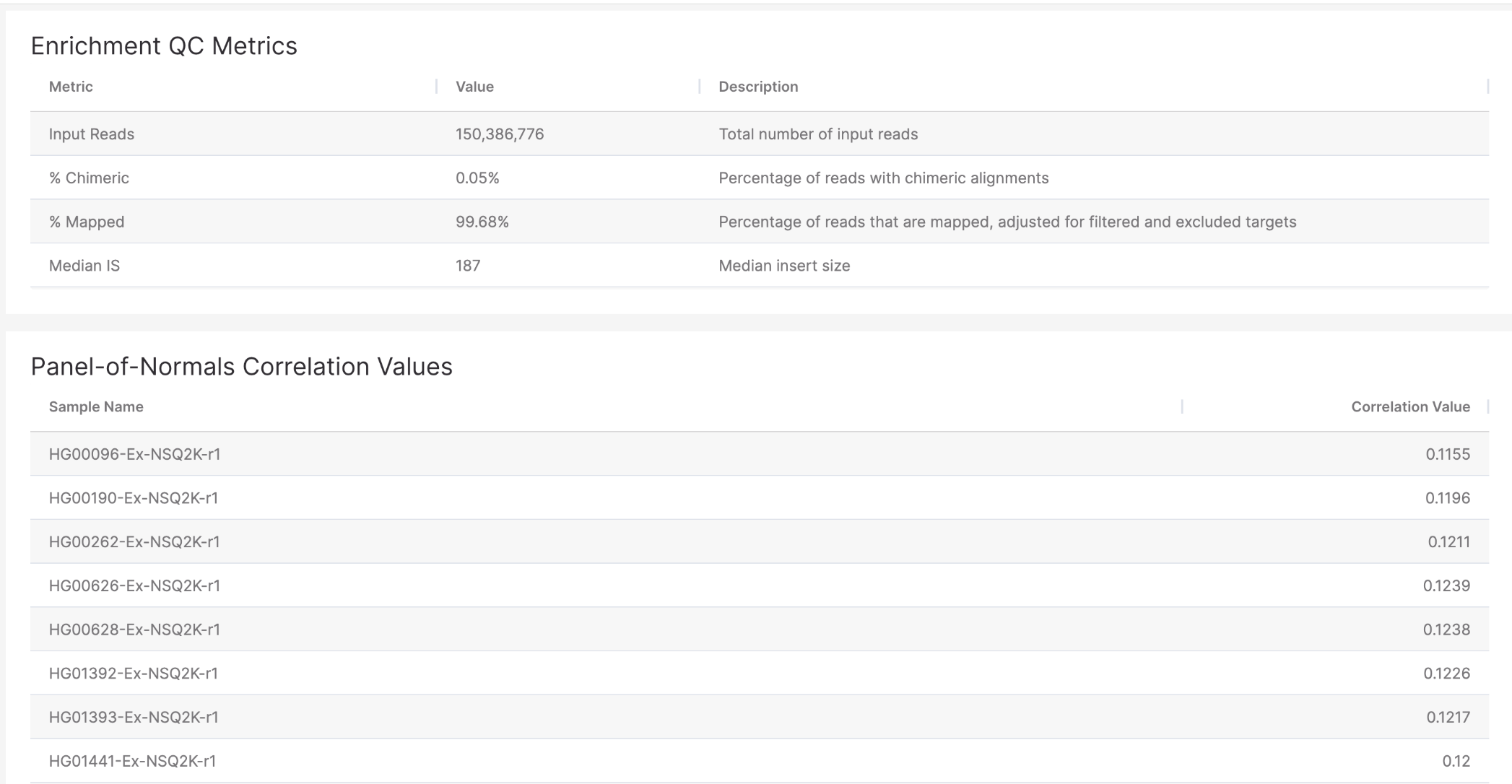
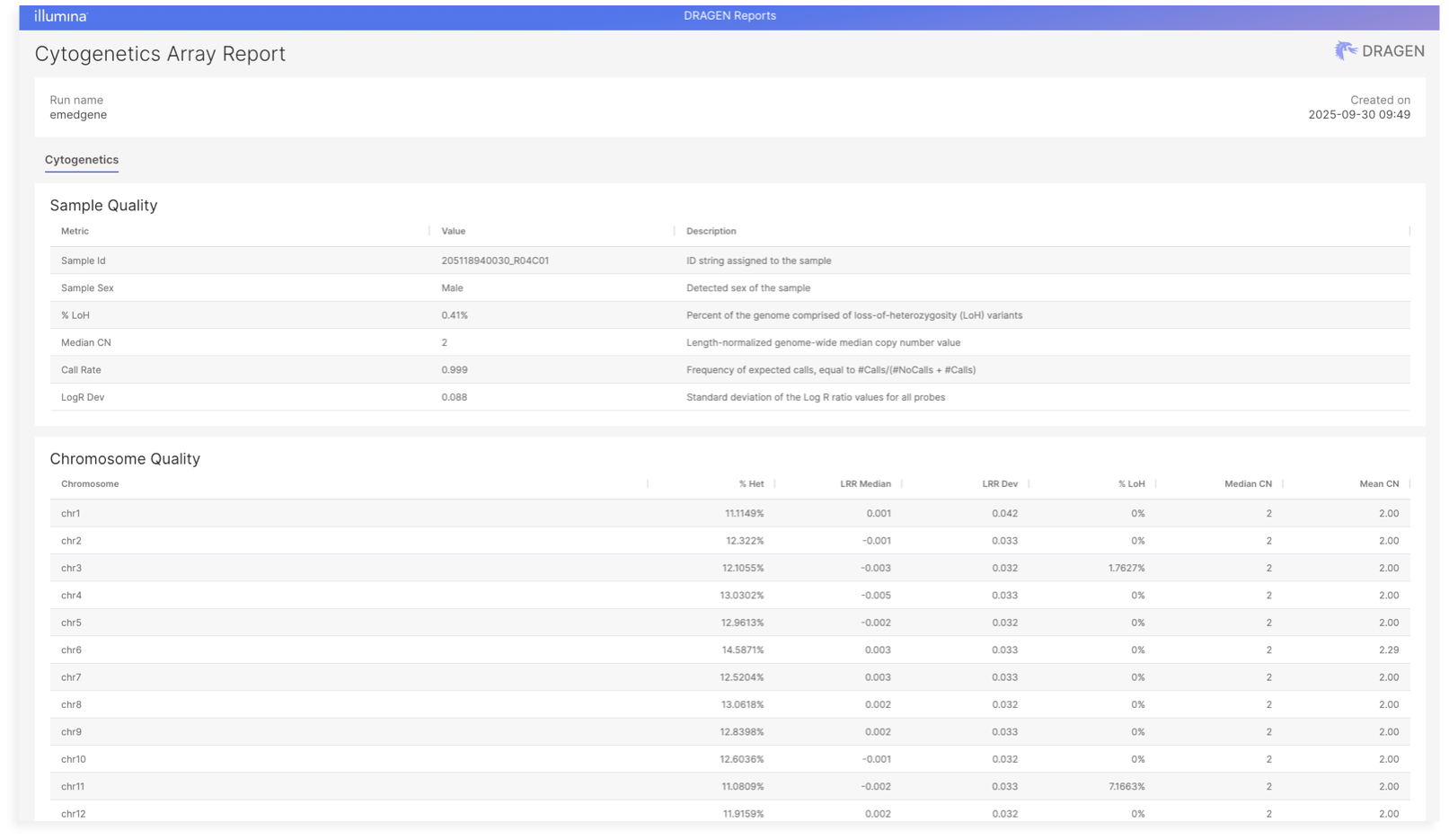
DRAGEN reports (accessible via a hyperlink in the Emedgene lab tab) now include a Panel-of-Normals (PON) Correlation section under the QC tab. This new table displays correlation values between the analyzed sample and a set of normal reference samples, helping assess how closely the sample resembles typical, non-pathogenic profiles.
Each row lists a reference sample ID along with its corresponding correlation score. This information can support quality control and help flag potential outliers or unexpected patterns. The table is also available in the downloadable DRAGEN report for each sample from the cases Lab page in Emedgene.
Example report:
### New! Support for DRAGEN Ploidy caller
Starting with Emedgene V100.39.0 Emedgene supports ingestion of the DRAGEN ploidy caller in addition to the ploidy estimation metrics. This support will cover DRAGEN 4.2, 4.3 and 4.4 (from VCF only).
\
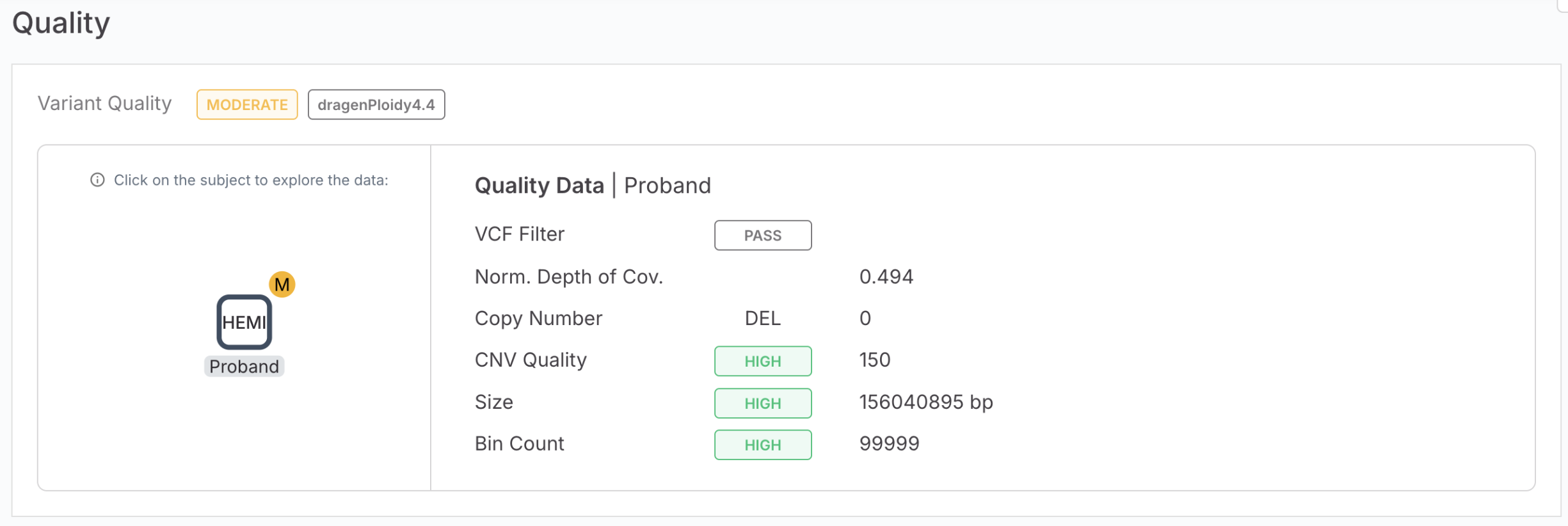
The Variant Page now displays an additional quality metric to help you better assess Copy Number Variant (CNV) results from the Ploidy caller.
A new quality field called "Norm. depth of cov." is now available in the Quality tab of the Variant Page. This metric provides valuable insight into the sequencing coverage depth for CNV variants, helping you make more informed quality assessments. This new quality field appears in the Variant Page | Quality tab | Variant quality card directly after the VCF FILTER information.
Values are automatically calculated and displayed with up to 3 decimal places for precision.
The field only appears when data is available for the specific variant. If no normalized depth of coverage data exists for a variant, the field will not be shown.
### New! Support for HLA genes from targeted.json
Starting with Emedgene V100.39.0 and DRAGEN 4.4, Emedgene will support the ingestion of the HLA genes from the targeted.json. Interpretation of this data will follow existing haplotype capabilities for CYP2D6, CYP2B6 and star allele caller outputs.
{% hint style="info" %}
Limitation: The following HLA genes are not supported – HLA-R, HLA-DRB3, HLA-DRB4, HLA-Y.
{% endhint %}
### DRAGEN 4.4 SV caller updates
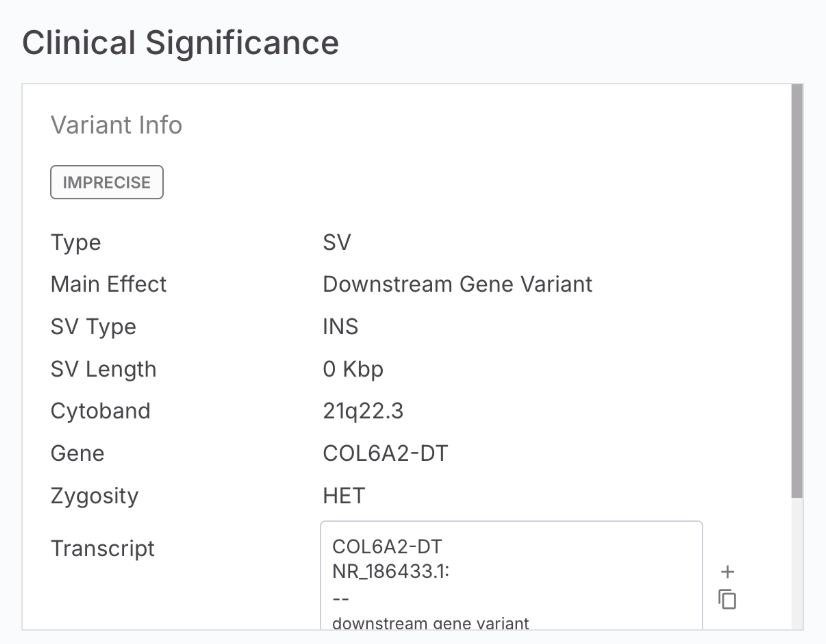
Starting with Emedgene V100.39.0 and DRAGEN 4.4, Emedgene will display the Imprecise tag on SV caller outputs.
The IMPRECISE tag indicates that:
* The variant's breakpoint locations are approximate rather than exact.
* Additional consideration may be needed when interpreting the variant's genomic coordinates.
* The variant calling algorithm was unable to determine precise breakpoint boundaries.
In Emedgene, the Imprecise tag will be displayed as a chip in the Variant Page | Clinical Significance tab | Variant Info card, similar to other tags such as Mosaic.
### Quality metrics for DRAGEN 4.4
Variant Type/Caller
Emedgene Quality Calculations
SNV and small InDels
A variant is designated as Low quality if:
· The VCF/FILTER is not "PASS" OR the VCF/QUAL is less than 10
A variant is designated as High quality if:
· The VCF/FILTER is "PASS" AND the VCF/QUAL is greater than 30
All other variants are categorized as Moderate quality.
The VCF Filter value will be presented in the Variant Page | Quality tab.
CNVs
(called by the CNV_SV caller for genomes)
A variant will be designated as Low quality if:
· The VCF/FILTER is not "PASS" AND INFO field SVCLAIM = D
A variant will be designated as High quality if:
· The VCF/FILTER is "PASS" AND (INFO field SVCLAIM = D OR INFO field SVCLAIM = DJ) AND QUAL > 100.
All other variants are categorized as Moderate quality.
CNVs
(called by read-depth caller)
A variant will be designated as Low quality if:
· The VCF/FILTER is not "PASS"
A variant will be designated as High quality if:
· The VCF/FILTER is "PASS" AND VCF/QUAL is greater than 30
All other variants are categorized as Moderate quality.
CNV ASCN
(whether output is in the CNV_SV caller or the read-depth caller)
A variant will be designated as Low quality if:
· The VCF/FILTER is not "PASS"
A variant will be designated as High quality if:
· The VCF/FILTER is "PASS" AND VCF/QUAL is greater than 40
All other variants are categorized as Moderate quality.
CNV ASCN with Cytogenetics Modality
A variant will be designated as Low quality if:
· The VCF/FILTER is not "PASS" or the event has a “SEGID” tag
A variant will be designated as High quality if:
· The VCF/FILTER is "PASS" AND VCF/QUAL is greater than 40
All other variants are categorized as Moderate quality.
SVs
A variant will be designated as Low quality if:
· The SVLEN is greater than 50 kb OR the VCF/FILTER is not "PASS"
A variant will be designated as High quality if:
· The VCF/FILTER is "PASS" AND VCF/QUAL is greater than 500
All other variants are categorized as Moderate quality.
Ploidy
A variant will be designated as Low quality if:
· The VCF/FILTER is not "PASS"
A variant will be designated as Medium quality if:
· The VCF/FILTER is "PASS"
STR
A variant will be designated as Low quality if:
· The VCF/FILTER is not "PASS"
A variant will be designated as High quality if:
· The VCF/FILTER is "PASS"
Additional STR loci will always have low quality: ARX, HOXA13
MRJD
A variant will be designated as Low quality if:
· The VCF/FILTER is not "PASS"
· The VARIANT is in “unreliable_loci.vcf” AND JGT is 0/0/0/1 or 0/0/1/1
· The VCF/FILTER is “PASS” AND INFO contains “MRJD_HS;REF_DIFF” AND FORMAT/GT = (0/0/0/1 OR 0/0/1/1 OR 0/0/0/0/0/1 OR 0/0/0/0/1/1 OR 0/0/0/1/1/1 OR 0/0/1/1/1/1)
A variant will be designated as High quality if:
· FILTER=PASS and INFO contains (“UNIQUELY_PLACED” OR “REGION_AMBIGUOUS” OR “MRJD_HS;ALT_LOCATION”)
All other variants are categorized as Moderate quality.
Note: Unreliable loci are sites where a common population allele in the pseudogene would be a pathogenic variant if that were present in the active gene.
In such cases, we would very frequently have a JGT of 0/0/0/1 or 0/0/1/1 due to the ALT allele being frequently present in the pseudogene, and that site would always be annotated as pathogenic in the gene position. Therefore, we mark that locus as low quality unless it's present in at least 3 copies of the segdup.
Targeted
A variant will be designated as Low quality if:
· Filter is not PASS
A variant will be designated as High quality if:
· FILTER=PASS
Star Alleles
All variants (star allele, CYP2D6, CYP2B6, HLA genes) are designated as High quality.
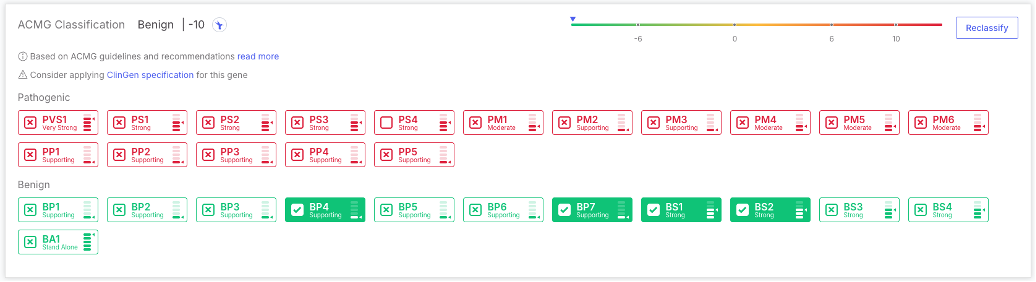
## ACMG updates classification module for SNVs
Our most requested feature update by customers has been to annotate this module with gene-specific ClinGen VCEP guidelines, and this has been completed in this release. (If you aren’t yet voting on Emedgene feature requests, they can be accessed from the navigation bar, underneath the help center link. We are prioritizing most-voted feature requests in every release.)
We have also completed the update of our automated ACMG classification module to the latest guidelines according to the table below. All updates to this module have taken into account the upcoming SVCv4 guidelines, and will help accelerate implementation of the new guidelines once those are released.
Specifically in this release we have updated the calculations for PS1, BS2, PP1/BS4, BP5, and added the possibility to exclude PP5/BP6 from the auto-calculation. All tags include improved evidence.
| Tags | Guidelines | Note | Version |
| --------------------------------- | ----------------------------- | ------------- | -------- |
| PVS1 | Abou Tayoun 2018; Walker 2023 | SpliceAI 10k | V38 |
| PS1 | Walker 2023 | | V39 |
| PS2/PM6 | SVI 2021 | | V38 |
| PS3/BS3 | Walker 2023; Brnich 2020 | | V38 |
| PS4/BS2 | | | V39 |
| PM1, PM4/BP3, PM5, PP2, BP1, BP2 | Up-to-date | | N/A |
| PM2/BS1/BA1 | SVI PM2, Gosh 2018 | | V37 |
| PM3 | SVI PM3 2019 | | V38 |
| PP4 | Biesecker 2024 | | V38 |
| PP1/BS4 | Biesecker 2024 | | V39 |
| PP3/BP4 | Walker 2023; Pejaver 2022 | | V37 |
| PP5/BP6 | Biesecker 2018 | | V39 |
| BP5 | Biesecker 2018 | | V39 |
| BP7 | Walker 2023 | | V38 |
### ClinGen VCEP annotation and links to gene-specific classification considerations
We have expanded the ACMG classification experience by integrating gene-specific guidelines from the ClinGen Variant Curation Expert Panels (VCEPs), providing deeper alignment with expert-reviewed standards.
In the ACMG Classification card, variants located within genes that have a released ClinGen specification are now automatically annotated. A dedicated note with an icon is displayed, linking directly to the relevant ClinGen recommendation page at . This enhancement builds on the existing ACMG visual framework by adding a clear, actionable reference to gene-specific classification considerations.
If a variant requires manual review based on these specifications, an alert will notify the user, ensuring that expert guidance is applied where needed.
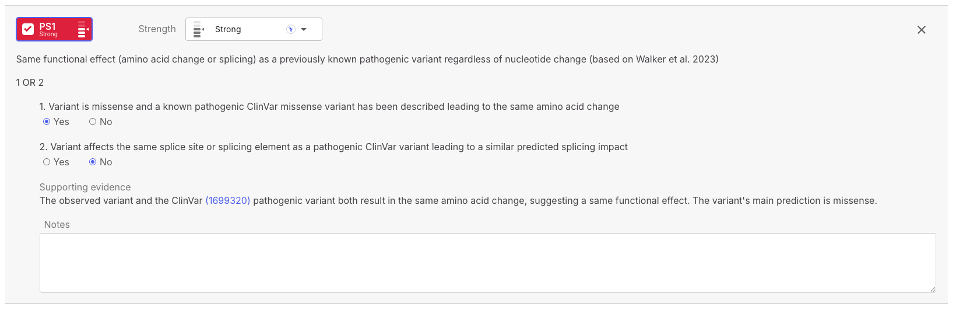
### PS1 updated according to Walker *et al.* 20232 with improved evidence
PS1 is an ACMG/AMP criterion applied when a variant results in the same functional effect, either amino acid change or splicing impact, as a previously known pathogenic variant, regardless of nucleotide change.
In this version, we refined the logic to support both missense and splice variants. For missense, PS1 is applied when the variant leads to the same amino acid substitution as a ClinVar pathogenic variant on the same codon. For splice variants, PS1 is applied when the variant affects the same splice site or region as a ClinVar pathogenic variant, with similar predicted impact based on SpliceAI and SpliceAI-10K.
Strength is assigned based on variant type, location, and ClinVar classification. Supporting evidence includes links to the ClinVar variant, SpliceAI score, and predicted effect.
To prevent over-weighting, PS1 is excluded from ACMG classification when assigned Supporting strength and matched to a Likely Pathogenic variant. Additionally, When PS1 is used with PVS1 at Very Strong strength, the combined contribution is capped at Very Strong + Supporting (Walker *et al.* 20232).
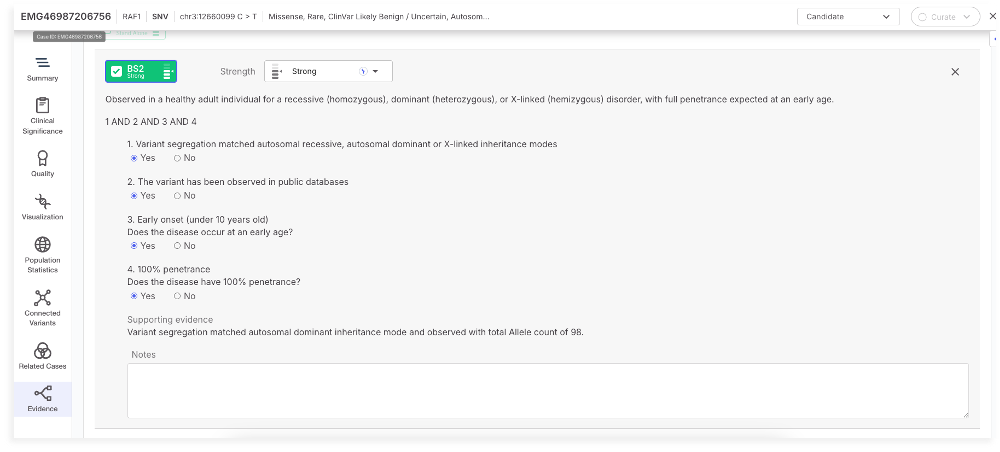
### BS2
The BS2 tag supports a benign classification when a variant is observed in healthy individuals for a disorder with full penetrance expected at an early age. This update introduces a structured logic framework to ensure consistent and guideline-compliant application.
The automated ACMG classification module for SNVs has updated logic for assigning the BS2 criterion.
The updated logic now applies a four-part decision framework to determine BS2 activation based on inheritance mode, population frequency, disease onset, and penetrance. These conditions are evaluated using curated inheritance data, public variant databases, and manual review inputs.
BS2 is positively activated when all four conditions are met: the variant’s segregation pattern matches a known inheritance mode (AD, AR, or XLR). The variant is observed in public databases at a frequency inconsistent with full penetrance, and the disease is both early-onset and fully penetrant. Supporting evidence includes the observed inheritance mode and the relevant count from public databases, providing users with clear justification for classification.
### PP1/BS4 updated according to Biesecker *et al.* 20245
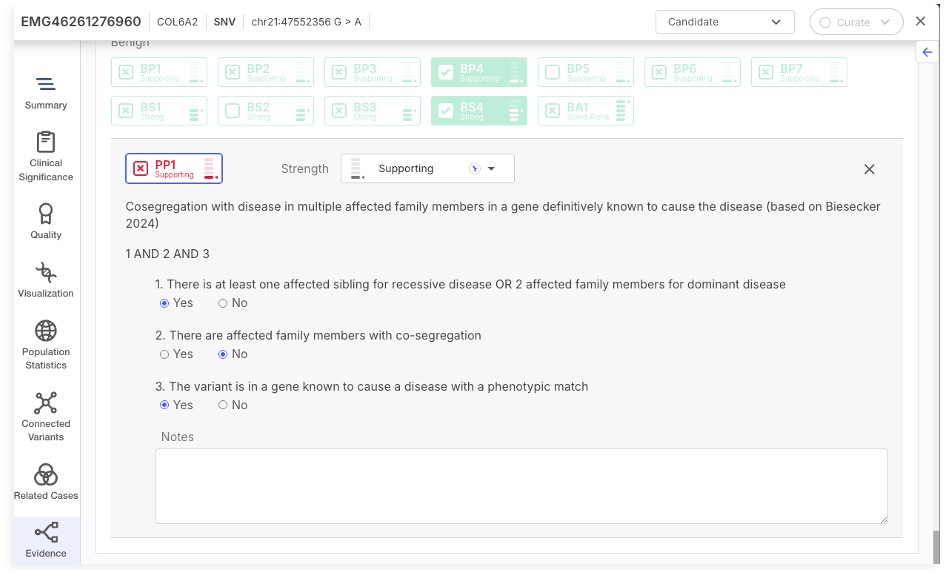
PP1 and BS4 are ACMG/AMP criteria based on segregation evidence. PP1 is applied when a variant co-segregates with disease in multiple affected family members, supporting pathogenicity. In contrast, BS4 is used when a variant fails to segregate with disease in affected individuals, supporting a benign classification.
In this version, we provide an updated classification framework for both PP1 and BS4 based on pedigree structure. Inheritance mode, and phenotypic match. For PP1, activation requires the presence of affected relatives, co-segregation of the variant with disease, and a phenotypic match. Strength is calculated based on the number of affected and unaffected family members and mapped to Supporting, Moderate, or Strong.
BS4 is activated when there is at least one additional affected family member and the variant does not segregate with disease, based on genotype mismatch with expected inheritance. BS4 is assigned a Strong evidence level by default.
Supporting evidence for both tags includes the selected gene, disease, inheritance mode, and number of affected/unaffected individuals relevant to the segregation pattern. If coverage data is incomplete, PP1 will display a message indicating that co-segregation cannot be determined.
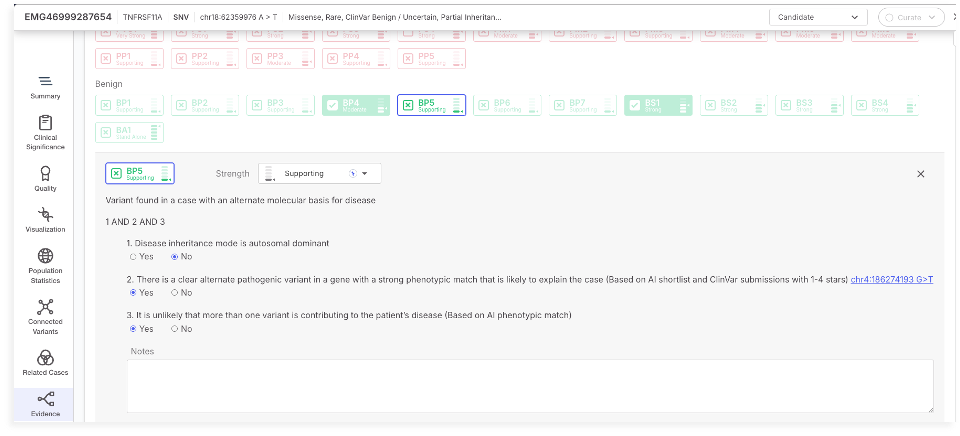
### BP5 updated according to Biesecker *et al.* 20187
The BP5 tag is used to support a benign classification when a variant is found in a case with an alternate molecular basis for disease. This means that another variant in the same case is more likely to explain the observed phenotype, reducing the likelihood that the variant under evaluation is contributing to the disease.
The automated ACMG classification module for SNVs has updated logic for assigning the BP5 criterion.
BP5 is positively activated when all three conditions are met: the disease is autosomal dominant, a second variant with strong pathogenic evidence and phenotypic match is present, and the AI model indicates that a single variant is likely responsible for the disease. Supporting evidence includes the alternate variant’s identity, gene, and phenotypic score, providing users with transparent context for the classification.
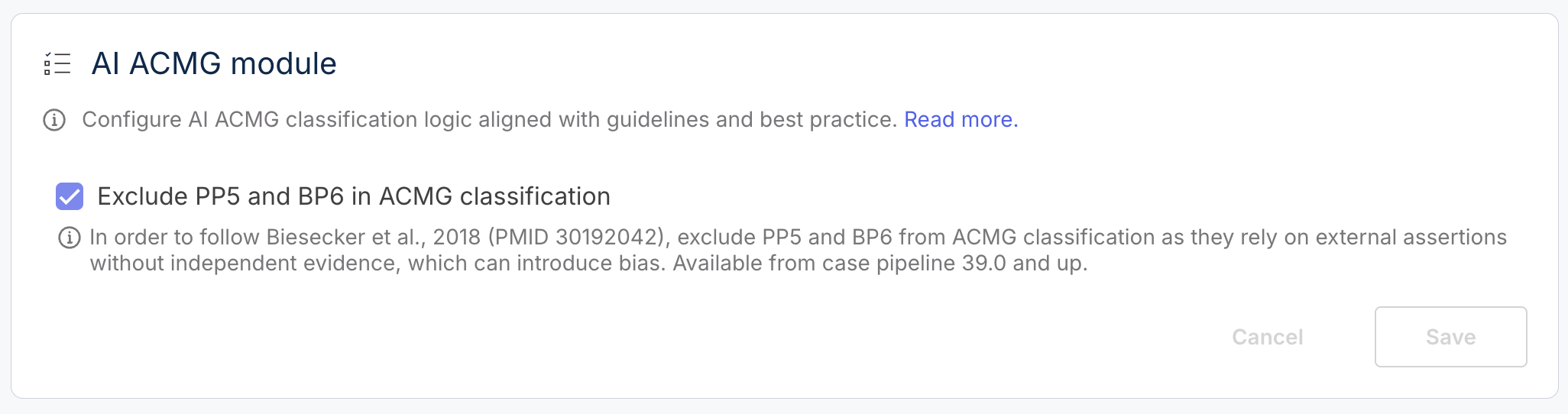
### Opt to exclude PP5/BP6 from the overall calculation
PP5 and BP6 are ACMG/AMP criteria based on external assertions. PP5 supports pathogenicity when a reputable source reports a variant as pathogenic, while BP6 supports benign classification when a source reports a variant as benign. However, both tags rely on external claims without independent evidence, which can introduce bias.
In this version, no changes were made to the logic or questions for PP5 or BP6. Instead, we introduced a new configuration option in the Organization Settings that allows users to exclude these tags from ACMG classification. This update aligns with recommendations from Biesecker *et al.* 2018, which caution against using externally asserted classifications without supporting data.
By default, PP5 and BP6 are excluded from ACMG classification. When excluded, a warning message is displayed in the variant page if either tag is positive. Users with the new ‘manage AI ACMG’ role can toggle this setting and save changes. All updates to this configuration are logged with username and timestamp.
This feature is available starting from case pipeline version 100.39.0 and ensures greater transparency and control over how external assertions are handled in variant interpretation workflows.
References:
* *Abou Tayoun, A. N., Pesaran, T., DiStefano, M. T., Oza, A., Rehm, H. L., Biesecker, L. G., ... & ClinGen Sequence Variant Interpretation Working Group (ClinGen SVI). (2018). Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Human mutation, 39(11), 1517-1524.*
* *Logan C Walker, Miguel de la Hoya, George A R Wiggins et al. Using the ACMG/AMP framework to capture evidence related to predicted and observed impact on splicing: Recommendations from the ClinGen SVI Splicing Subgroup. Am J Hum Genet. 2023 Jul 6;110(7):1046-1067. PMID: 37352859.*
* *Brnich, S. E., Abou Tayoun, A. N., Couch, F. J., Cutting, G. R., Greenblatt, M. S., Heinen, C. D., ... & Berg, J. S. (2019). Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome medicine, 12(1), 3.*
* *Ghosh, R., Harrison, S. M., Rehm, H. L., Plon, S. E., Biesecker, L. G., & ClinGen Sequence Variant Interpretation Working Group. (2018). Updated recommendation for the benign stand‐alone ACMG/AMP criterion. Human mutation, 39(11), 1525-1530.*
* *Biesecker, L. G., Byrne, A. B., Harrison, S. M., Pesaran, T., Schäffer, A. A., Shirts, B. H., ... & Rehm, H. L. (2024). ClinGen guidance for use of the PP1/BS4 co-segregation and PP4 phenotype specificity criteria for sequence variant pathogenicity classification. The American Journal of Human Genetics, 111(1), 24-38.*
* *Pejaver, V., Byrne, A. B., Feng, B. J., Pagel, K. A., Mooney, S. D., Karchin, R., ... & Topper, S. (2022). Calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for PP3/BP4 criteria. The American Journal of Human Genetics, 109(12), 2163-2177.*
* *Biesecker, L. G., & Harrison, S. M. (2018). The ACMG/AMP reputable source criteria for the interpretation of sequence variants. Genetics in Medicine, 20(12), 1687-1688.*
## New and improved Variant Page external links to Google Scholar, PubMed, GeneCC & more
In response to customer feedback, we’ve significantly improved our Variant Page external links in this version, making it easier to identify relevant literature for interpretation and curation and adding new gene and gene-disease links.
Literature searches are now easier and more comprehensive with Google Scholar, PubMed and LitVar searches for SNV, Indel, mtDNA, CNV and SV variant types, in addition to the existing Genomenon link.
There is a new set of gene links to ClinGen, Decipher, GeneCC and OMIM. GeneCC gene-disease connections have also been added to the Gene-Disease relations, including classification strength. Finally, the Monarch Mondo ID is now displayed for each disease, including the link out and API availability.
### New literature search variant links
Located in the Variant page | Clinical Significance tab, under the ‘Resources’ field in the Variant info card. These search links offer several alternatives for variant-specific literature searches to ensure comprehensive coverage of the literature.
The search query construction has taken into account the [ClinGen Biocurator Working Group Guidelines](https://clinicalgenome.org/tools/educational-resources/general-curation-topics/literature-searching-and-annotation/) and current literature on genomic variant information available within publications in order to construct the most comprehensive searches for each variant type and resource.
The query logic is as follows across literature sources:
* Small variants: RS ID or HGVS coding change (e.g. c.215C>G) or and protein change (e.g. p.Arg72Gly), applied across affected transcripts and aliases, along with the gene name.
* CNVs: ISCN notation or cytoband or location and standard CNV descriptors and gene-level information.
* SV insertions: Cytoband or variant details and gene context and standard SV descriptors.
#### Google Scholar search link
The new Google Scholar search link is available for: SNVs, Indels, mtDNA, CNV deletions and duplications and SV insertion variants.
#### PubMed search link
The new PubMed search link is available for SNVs, Indels, mtDNA, CNV deletions and duplications and SV insertion variants.
Note: Analysis shows that searching PubMed and PMC (PubMed Central) using protein change (p.) values with single-letter amino acid notation retrieves approximately 44.95% of all relevant literature articles. To improve coverage, variant search links now include an additional query using the single-letter amino acid format in p. notation.
If no structured query is defined for a variant type, the link defaults to the main gene associated with the variant, or in cases of multiple genes, the one used for gene metrics.
#### LitVar2 search link
A new LitVar2 search link has been added to the ‘Resources’ field under the Variant Info section (Clinical Significance tab) on the variant page, enabling users to search for variant-specific literature.
LitVar2 is an advanced literature searching tool developed by NCBI that connects variants to biomedical publications. Using enhanced text mining, natural language processing, and name normalization, it enables accurate retrieval of variant-specific information and returns consistent results across different variant representations.
LitVar2 search links are available for small variants only (SNVs and Indels).
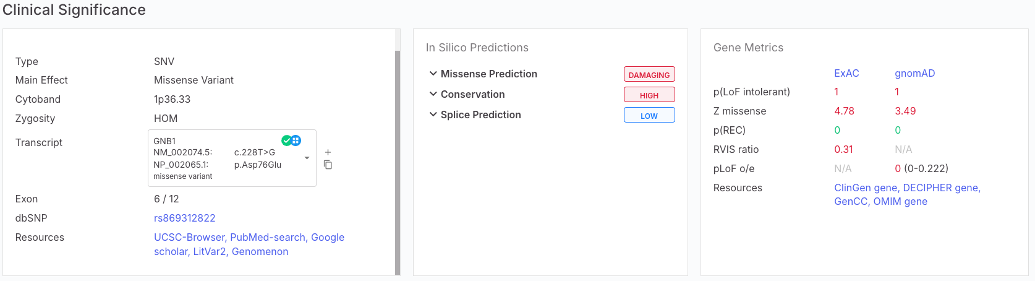
### New gene resources links
A new set of gene resources links is now available on the Variant Page→ Clinical Significance tab, within the newly added ‘Resources’ field under the Gene Metrics card.
These resource links provide direct access to databases that provide detailed information about gene function, associated diseases, inheritance patterns, and clinical relevance. This addition enhances the interpretive context and complements variant-level literature review tools.
Included resources:
* OMIM (Online Mendelian Inheritance in Man):
* GenCC (Gene Curation Coalition):
* ClinGen (Clinical Genome Resource):
* DECIPHER:
* gnomAD: (Genome Aggregation Database):
Gene-level links are available for the following variant types:
* Small variants (SNVs, small indels, including mitochondrial DNA)
* STR variants
* SV variants
For mtDNA and STR variants, a dedicated gnomAD gene-level link is included.
External links to these resources are presented for the main gene associated with the variant. If the variant affects multiple genes, links are shown for the gene used in the Gene Metrics card.
As part of this update, GeneCards and WikiGenes external links have been removed for all variant types to improve clarity and emphasize high-confidence sources. These resources are no longer supported in the Variant Info card under the Clinical Significance tab.
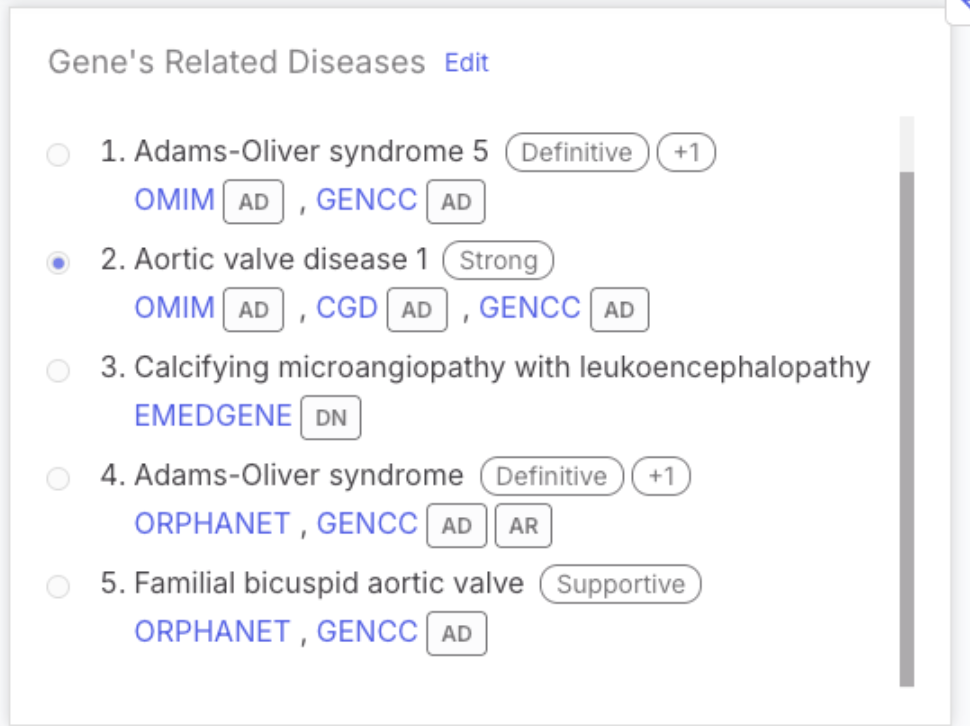
#### GenCC gene-disease relationships and classification strength
The Gene Curation Coalition (GenCC) provides standardized gene-disease relationship (GDR) classifications from multiple expert submitters. Unlike many resources that use inconsistent terminology, GenCC enables more reliable GDR assessments by unifying evidence definitions across contributors. Emedgene’s Knowledge Graph now includes the GDR, classification strength, submitter name, submitter's GenCC ID, date of submission/evaluation and inheritance mode as reported by submitter. The GeneCC GDR resource will be updated on a monthly cadence with the rest of the Emedgene Knowledge Graph resources.
The GDR links to GenCC will be displayed in:
* Variant Page |Summary Tab | Gene-Related Diseases
* Variant Page | Clinical Significance | Gene Metrics | Resources (New)
The GDR information will also be displayed on the Variant Page | Gene’s Related Diseases.
The card will display:
* Associated diseases for the variant’s gene
* Inheritance mode aggregated from all submitters
* Most supportive classification (e.g. Definitive), followed by a count of additional classifications (e.g. Definitive +2)
* A hover tooltip that reveals a breakdown of all classifications and the names of submitters who provided them
GenCC is listed last in the source priority order, following OMIM, EMEDGENE, CGD, and Orphanet.
#### Monarch Initiative MONDO links
The Monarch Initiative is an open-science project that integrates genotype–phenotype data across species using semantic technologies to support disease diagnosis, research, and discovery. The initiative also develops and maintains MONDO, a unified disease ontology that harmonizes disease definitions across multiple sources to enable consistent and computable disease representation.
Monarch MONDO links are displayed in the Variant Page | Summary tab | Gene-Related Diseases.
When evaluating a variant and its associated gene-disease relationships, a Monarch external link is shown for eligible diseases in the Gene-Related Diseases section. This link redirects to the corresponding MONDO entry on the Monarchy Initiative database.
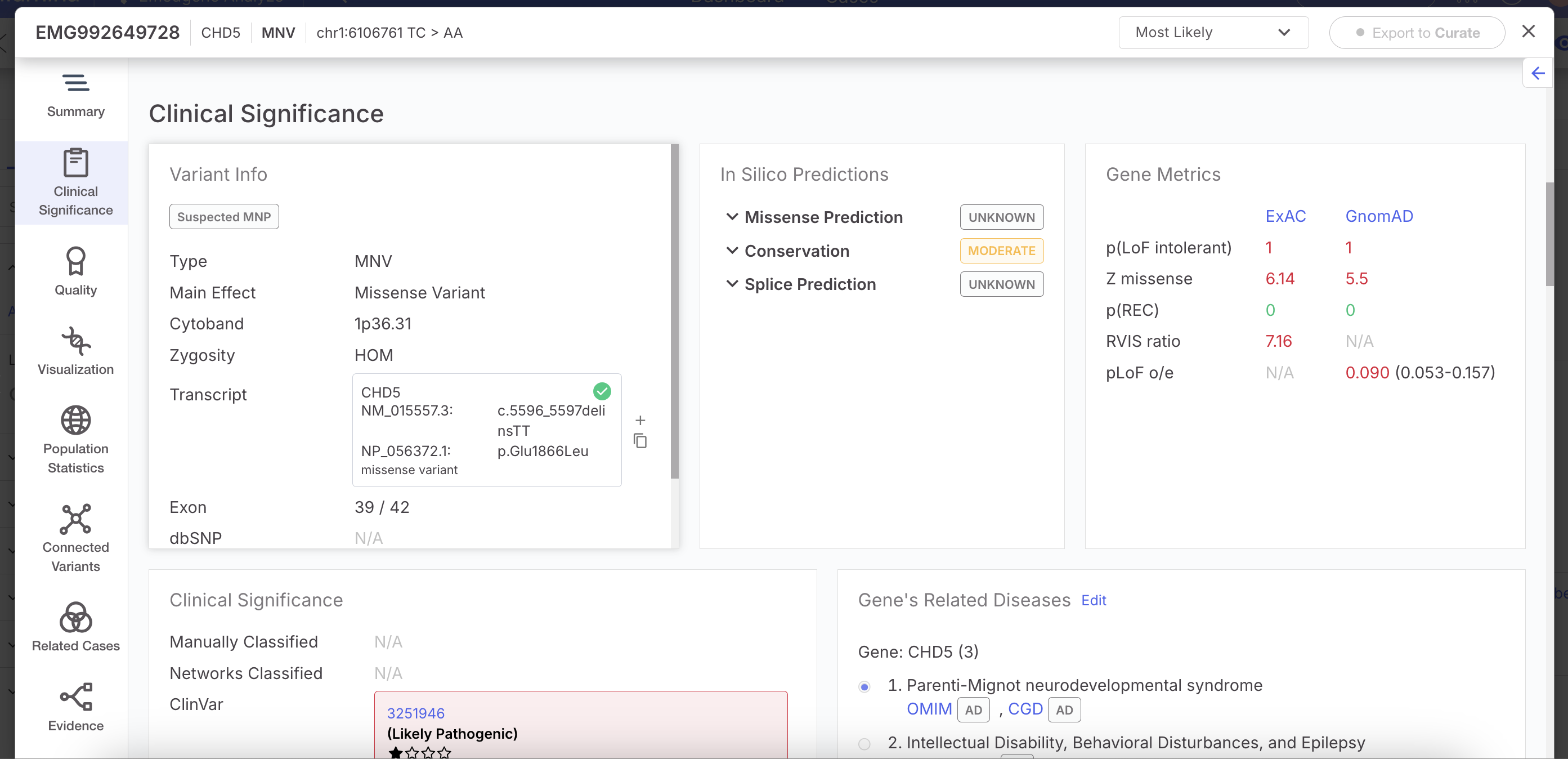
## Improved Support for Multi-Nucleotide Variants (MNVs)
Emedgene now supports ingestion, annotation, and filtering of Multi-Nucleotide Variants (MNVs), a class of genetic variants where two or more adjacent nucleotides are substituted simultaneously. This enhancement improves biological accuracy and clinical relevance, especially when nearby substitutions affect the same codon or reading frame.
#### VCF ingestion, normalization and annotation
MNVs are now properly recognized during VCF ingestion and stored in two forms:
* New: As a single combined variant (e.g. AG>TC), representing the full multi-nucleotide change.
* Existing: As individual SNVs (e.g. A>T and G>C), for compatibility with existing tools and workflows.
MNVs are now annotated both as combined variants and individual SNVs, Annotations include transcript effect, population frequency, and prediction scores, powered by trusted sources such as VEP, GERP, and ClinVar.
#### Filtering and preset filters
MNVs are now available in the ‘Variant Type’ filters, are supported across all filterings including Quality. They can also be searched directly using REF/ALT base combinations.
#### Variant Page for MNVs
MNVs are now shown as a distinct variant type across the Variant Page, using the same layout as SNVs. Key updates include:
* Variant Summary and Variant Info: Main effect, transcript, cytoband, zygosity, exon number and dbSNP.
* Quality section: VCF filter, base quality, read depth, mapping quality, and genotype quality, with visual indicators (High/ Moderate/ Low).
* Connected Variants tab will group MNVs with adjacent SNVs.
* Related Cases table will include MNVs when matching chromosome, position, REF, and ALT.
#### Interpretation and reporting
MNVs are fully supported in interpretation workflows, including tagging, pathogenicity assignment and reporting/exporting.
{% hint style="info" %}
Limitations:
* Curate does not support MNVs yet and export to Curate is blocked.
* The AI shortlist does not support MNVs.
* ACMG classification is disabled for MNVs.
* MNV variants in mtDNA genes will not show up in a filter restricted to mtDNA variants
{% endhint %}
Note: MNV calling is not turned on by default in DRAGEN. More information can be found in the [DRAGEN 4.4 product guide](https://help.dragen.illumina.com/product-guide/dragen-v4.4/dragen-dna-pipeline/small-variant-calling#command-line-options-for-merging-phased-variants).
## Cross-Cloud Opt-In Data Sharing
The Emedgene Data Sharing Networks has been supported within cloud region since V32.0. This capability empowers trusted partners to securely share variant information beyond allele frequency, including pathogenicity, phenotypes, ACMG tags [and more](https://help.emg.illumina.com/emedgene-analyze-manual/analyze_network/network_sharing_configuration). Through a flexible, case-based consent model, collaborating organizations can establish multiple networks with granular control over data sharing levels, configuring the data sharing per network. Labs retain full control over their sharing preferences, supporting both broad collaboration and precise data governance. This architecture not only augments internal lab data but also fosters the democratization of genomic insights across institutions.
Starting in v39, labs can collaborate across cloud environments and geographic regions, expanding the scope of potential collaborations. The same secure, opt-in data sharing module is enabled across any Emedgene cloud.
All existing data sharing network components support cross region sharing.
* Analyze | Related Cases
* Curate | Related Cases
* Curate | Variant search – has a new selector for regions, as the search functionality is not available for all regions concurrently.
* Settings | Network – New AWS region and infrastructure selector
{% hint style="info" %}
Limitation:
* If two or more networks share identical workgroup/organization names (possible across cloud regions today), there will be no way to distinguish between them in the UI. Recommendation to ascertain uniqueness of network naming across collaborators.
* Network classification no longer appears on the Variant Page | Clinical Significance tab or in the Analysis Tools table.
{% endhint %}
## CNV and Cytogenetic Interpretation Flow Improvements
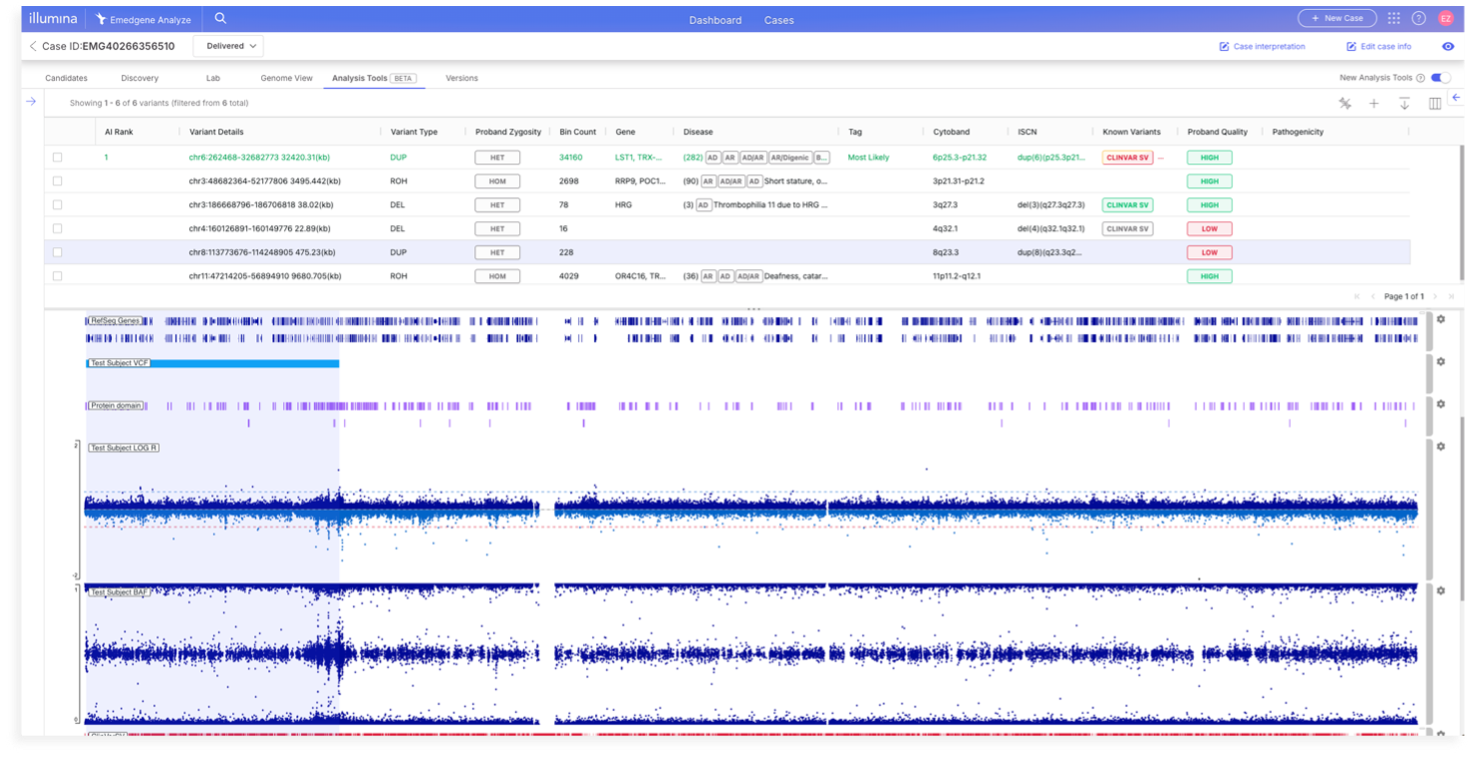
We're excited to introduce the beta version of our new Analysis Tools tab, which adds the ability to jointly view the analysis tools and a full IGV visualization in a single combined tab. This supports cytogeneticists’ need to view both structured variant data in a tabular format along with visual data in a single pane. New in this release is support for DRAGEN Array v1.3, which adds mosaic calling for Illumina cytogenetic arrays and overall improved CNV calling performance. We’ve also added new DRAGEN Array caller quality metrics.
### New combination analysis tools and visualizations (beta)
Users now have a streamlined flow for interpreting variants, with a unified view for both tabular variant details and full IGV visualization capabilities.
On the Analysis Tools tab, there is a New Analysis Tools toggle in the upper right. When turning it on, users will have access to the new tab which will show both the table and the visualization. The visualization is the same IGV available in the Variant Page | Visualization tab with the exact same functionality.
The display window height is draggable to adjust the visualization window for maximum review comfort. To turn off the visualization component, or back on again, a new visualizations icon has been added to the table navigation bar.
#### Table and visualization interaction
Some Analysis Tools table interactions are changed from the previous analysis tools. A single-click on a variant zooms the visualization to that region, whereas in the previous table it opened a variant page. To open a variant, use a double-click or the newly added ‘Open’ link on the left most side of the table. Once a variant is open, the arrow keys can still be used to navigate between variants.
If no variant is selected, the visualization component will display whole genome view (all chromosomes). Zooming in to a chromosome/variant level will display the more tracks.
Adjustment or merging of calls is performed via the ‘Add Variant’ button in the visualization component, in the same way it is enabled since V38.0 on the embedded IGV. When adjusting the span of the selected variant, the coordinates will be automatically populated to the Add Variant button.
### Support for DRAGEN Array V1.3
Emedgene provides an optimized interpretation and visualization solution for cytogenetic samples analyzed DRAGEN Array V1.3. Significant updates in [this release](https://help.connected.illumina.com/dragen-array/reference/release-notes/dragen-array-v1.3.0-release-notes) that are now supported in Emedgene:
* Mosaic calling and fraction estimation for mosaic events.
* Improved accuracy of sex chromosome calling, including pseudo-autosomal regions (PAR).
* New QC metrics are available in cytogenetics JSON output.
The following table summarizes the supported DRAGEN 1.3 compatibility:
| Type | DRAGEN Array Output | VCF (BYOD) | Notes |
| ----------- | --------------------------------------------------------------------------------------------- | ---------- | ----------------------------------------------------------------------------------------------------------------------------------------- |
| Cyto caller | \*cnv.vcf, \*baf.bedgraph, \*lrr.bedgraph, \*gt\_sample\_summary.json, \*annotated\_cyto.json | ✓ |
Visualization files are supported from batch upload/API only.
annotated\_cyto JSON is supported from DRAGEN Arrayv1.3 only.
|
### Visualizing and interpreting mosaic events
Every mosaic event will have a Mosaic tag, which appears on the Variant Page | Summary and Quality tabs. Users can create Preset Filters on this tag.
Additionally, the Variant Page | Quality Tab will display the Mosaic Fraction metric.
### Chromosome-level and sample-level quality metrics
DRAGEN Array V1.3 outputs a [cytogenetics JSON file](https://help.connected.illumina.com/dragen-array/product-guides/output-files#cytogenetics-annotation-json-file) that includes more sample-level, chromosome-level, and event-level metrics. When this file is ingested into Emedgene via the automated case creation flow, or a manual one, the quality metrics described below are available.
To view the new metrics, in the Quality Tab, each sample has a link to a DRAGEN report. The DRAGEN report will present:
Sample-Level Metrics
* Sample ID
* Sample sex
* Precent LOH
* Copy number median
* Call rate
* LogR deviation
Chromosome-Level Metrics
* Percent heterozygosity
* Log R Ratio (LRR) median and deviation
* Percent LOH
* Copy number median and mean
These metrics are displayed in the DRAGEN Report, accessible from the Sample Quality section of the Lab Page. You can also download the original quality files if provided during case creation.
## Additional Voice-of-Customer Improvements
#### Option to prioritize preferred transcript from Curate
To better support customers interested in leveraging their preferred transcripts from Curate, we have added the possibility to prioritize Curate transcripts as defined in Curate variants. This feature is only enabled for customers who turn on a new organization setting (accessible only via Illumina Bioinformatics support in this version).
The new transcript prioritization logic gives a higher priority to Curated variant transcripts, rank 3, in addition to Curate Gene rank 8.
1. VEP transcripts are prioritized over EFF transcripts.
2. If the case is a virtual panel, prioritize transcripts from genes in the case gene list (but not for Boosted Genes type panels).
3. Prioritize transcripts from Curate variants.
4. Prioritize RNA genes associated with disease (See [appendix 1](https://github.com/illumina-swi/emedgene-docs/blob/prod/docs/frequently-asked-questions/all-faq/how_does_emedgene_analyze_prioritize_transcripts/README.md#appendix-1-list-of-rna-genes-associated-with-disease) for prioritized list RNA genes). Importantly this does not apply to upstream and downstream RNA variants.
5. De-prioritize [biotype readthrough](https://www.ensembl.org/info/genome/genebuild/biotypes.html) transcripts.
6. Prioritize based on [impact](https://www.ensembl.org/info/genome/variation/prediction/predicted_data.html) in the following order: HIGH > MODERATE > LOW > MODIFIER.
7. Prioritize introns over UTR over upstream ([Appendix 2: MODIFIER effects prioritization)](https://github.com/illumina-swi/emedgene-docs/blob/prod/docs/frequently-asked-questions/all-faq/how_does_emedgene_analyze_prioritize_transcripts/README.md#appendix-2-modifier-effects-prioritization).
8. Prioritize organization canonical transcripts ([Defined in Curate](https://github.com/illumina-swi/emedgene-docs/blob/prod/docs/frequently-asked-questions/emedgene_curate_manual/curate_genes_2_28/curate_gene_page.md). Always applied, no settings needed).
9. Prioritize canonical transcripts (Based on [Appris](https://appris.bioinfo.cnio.es/#/)).
10. Prioritize transcripts from genes in the case gene list.
11. Prioritizing gene without “-” in their Name.
#### Improved structural variant merging logic
Improved handling for cases with structural variants originating from multiple samples. Starting in this version, structural variants will only be merged when they match based on Chromosome, Position, Reference, Alt and END. Prior to V100.39.0 the END position was not considered.
#### Genome pipeline speed down to 2:15 hours
We are continuously working to improve our genome pipeline speed for customers running rapid genomes or committed to quick turnaround times. In this release, a typical genome case should roughly 2 hours. This does not include the DRAGEN run time.
{% hint style="info" %}
Limitation: Customers accessioning via VCF+BAM can expect to experience 4-5 hour run time. We recommend using Joint gVCFs to experience the benefits of DRAGEN pedigree calling or accessioning with gVCFs.
{% endhint %}
#### Support for ACMG Secondary Findings v3.3
The ACMG secondary findings v3.3 gene list has been updated in both the XAI and the filters.
For the XAI, when a user selects to receive secondary findings, we will apply the v3.3 gene list, which includes 84 genes.
The All ACMG genes filter has also been updated to reflect this expanded list, including newly added genes: *PLN*, *ABCD1*, and *CYP27A1*.
This update ensures alignment with the latest ACMG recommendations for reporting clinically actionable secondary findings.
#### Updated default Emedgene Full Genes and Clinical Regions BED files
Emedgene default BED files have been updated with newly released regions from RefSeq\_curated (GCF\_000001405.40-RS\_2024\_08) and GenCode v47 to capture additional transcripts introduced in both hg38 and hg19.
This update also presents an opportunity to incorporate the latest ClinVar pathogenic variants and newly recognized RNA genes with disease associations. The new clinical regions (50 bp flanking) and full regions (5 kbp flanking) BED files have been merged with the previous versions to retain coverage of unique older regions. The coding files now focus solely on protein-coding regions (20 bp flanking) from both RefSeq\_curated and Gencode and includes selected RNA genes with disease relevance.
#### Self-Serve Enhancements
**Delete a variant in Curate**
Users now have the ability to delete entries directly from the Curate interface or via API, eliminating the need to contact support for corrections. From the UI, the delete functionality is accessed via the three dots menu. This functionality is available to users with the appropriate role and includes a confirmation step to ensure data is intentionally removed. Network-only variants, those originating from other organizations, remain protected and cannot be deleted. Once a variant is deleted, it is excluded from analysis pipelines and logged for audit purposes.
{% hint style="info" %}
Limitation: If the variant was used to annotate an existing case, the link from Analyze to Curate will be broken.
{% endhint %}
**Improved Preset management**
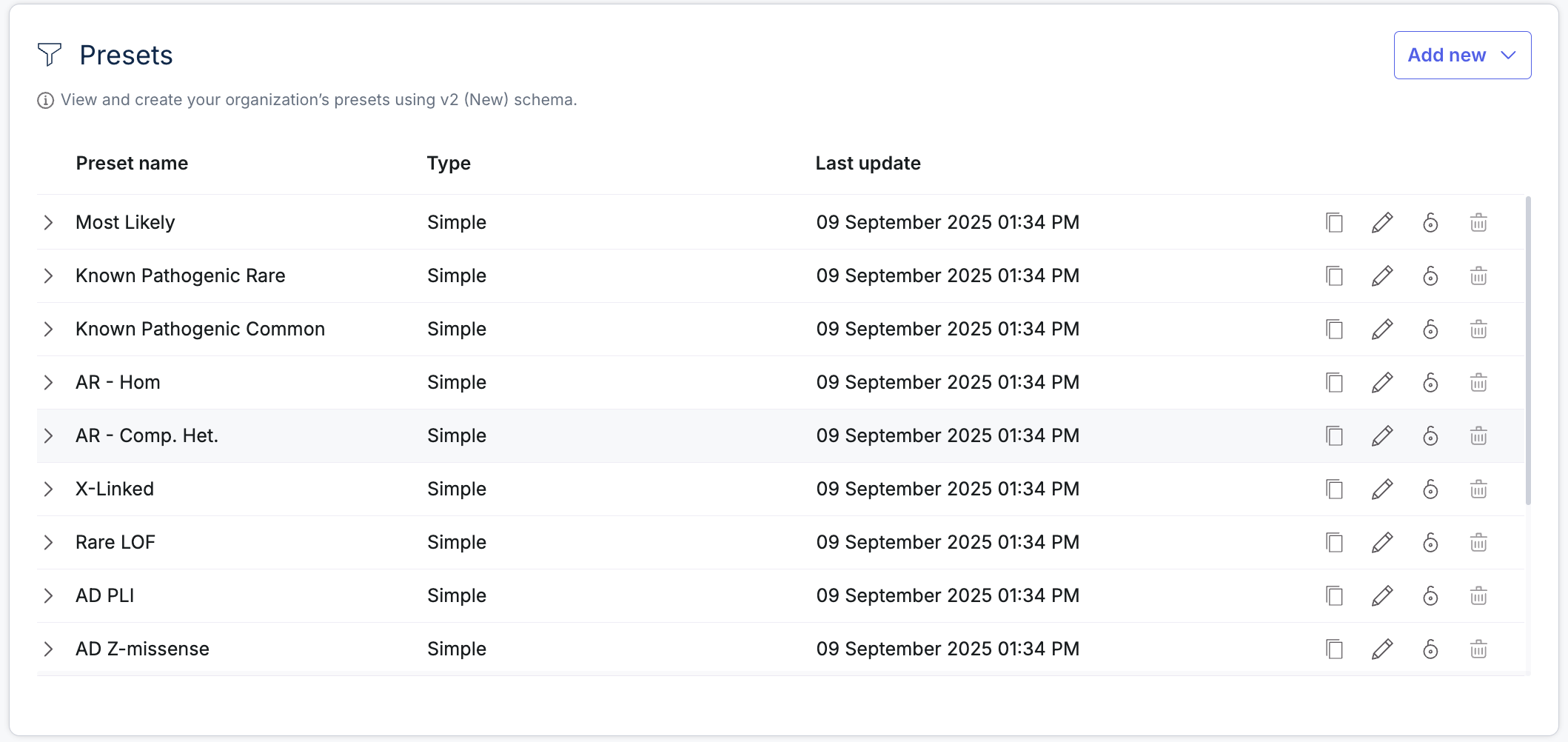
The preset management interface has been enhanced to better support users who rely on it frequently. Users can now duplicate existing presets, assign new names, and reuse filters with ease. A new column displays the last update for each preset, and sorting options have been added to organize presets by name, type, or update date. Additionally, preset names are now copyable across all tables, simplifying collaboration and reference.
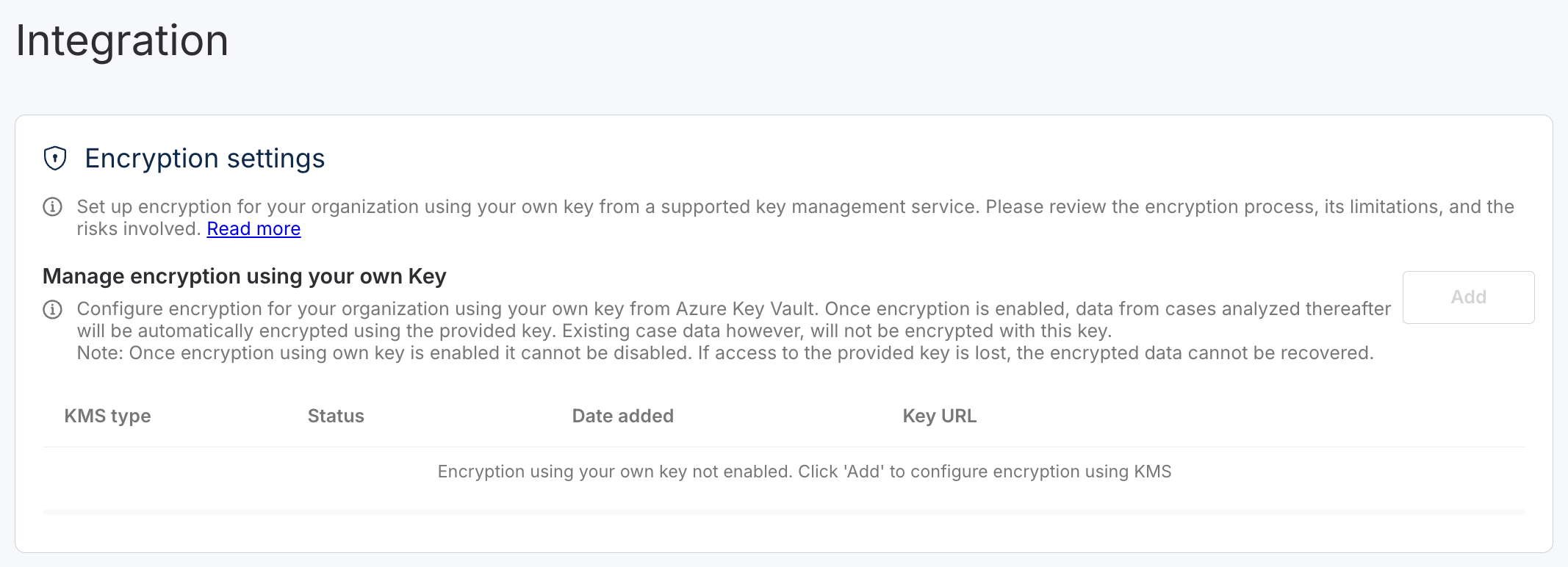
**BYOK integration**
Organizations can now configure encryption using their own key management services, commonly referred to as Bring Your Own Key (BYOK), directly within the Organization settings.
BYOK allows institutions to maintain full control over their encryption keys, supporting compliance with data protection regulations such as HIPAA and GDPR. The supported provider is Azure Key Vault. (AWS Key Management Service (KMS) will be added in subsequent versions).
## Limitations:
* Login | Emedgene does not support accents in User Names, despite support for these in IAM console. Users will not be able to login to the software.
* Add New Case | No validation that input files are uncorrupted, case will be created and fail.
* Add New Case | Selecting a disease should automatically suggest phenotypes, however, some diseases available for selection are from sources without phenotypes, and in that case, no phenotypes will be suggested.
* Add New Case | Adding metrics.tar.gz files is not supported from BSSH
* Add New Case | API/Batch/UI discrepancies:
* Cannot add phenotypes for unaffected parent in batch upload
* Cannot use the same gVCF file for multiple samples from the UI
* No validation for sample name in array JSON from batch upload/API
* Add New Case | BSSH | Human readable BSSH paths in batch upload do not work for some customers with large BSSH accounts.
* Add New Case | File name can be at most 255 characters.
* Pipeline | All samples with unknown sex are treated in the pipeline as female. Therefore, the presence of two X chromosome copies is treated as the reference (REF) condition. Customers can toggle on a ‘Keep ref variants’ setting to view these variants.
* Candidates, Variant Page, Curate | Evidence graph & ACMG automation will not be calculated for CNVs over 20MB. They will not have a gene related disease card in Curate.
* Lab Tab | Peddy contamination calculations may be inaccurate for panels due to small number of variants.
* Genome View | Only the largest 500 variants are displayed.
* Analysis Tools | Manually Added Variants | STRs | Format is not aligned with format of STRs on the software, e.g. missing variant length.
* Analysis Tools | Filters | DRAGEN SV caller contains a discrepancy between the VCF filter column and format field.
* Analysis Tools | Filters | ACMG pathogenicity filters don’t support CNVs.
* Analysis Tools | Filters | MNV variants in mtDNA genes will not show up in a filter restricted to mtDNA variants
* Analysis Tools | Presets | Preset filters v1 schema is deprecated, please upgrade to V2 prior to moving to any version over 37.
* Analysis Tools | Custom Presets & Settings | Presets – Cannot save custom and preset filters with {} in name.
* Analysis Tools | Search for CNVs by position does not consider end, only start.
* Candidates, Variant Page | After editing the evidence graph, phenotypic match strength indications are missing from the sidecar and variant page.
* Candidates | Evidence Graph | Changing the disease in the evidence graph will not automatically change the inheritance mode, that needs to be manually edited as well.
* Variant Page | Max AF in the analysis tools and export is across population DBs; the gnomAD card displays a different Max AF, referring only to gnomAD data.
* Curate | Pathogenicity sort does not sort by most to least pathogenic.
* Curate | Some CNV variants will not get a Network icon in the search despite having network variants.
* Webhooks | Cannot be trigger on internal software statuses such as ‘In Progress’ ‘Reanalysis’.
* Reporting | PMIDs will only work if there is an author on link, no support for books.
* Export to excel is limited to 32KB per cell, which may prevent exports with very large CNVs.
* Export/API | Applies a 6 digit rounding to the gnomAD AF, different than the 4-digit rounding in the UI.
* Organization Settings | BED upload | Validation on the UI component does not check the following. No validation at all for API uploads.
* All lines in the BED must contain the same number of columns.
* No duplicate lines.
* No trailing whitespaces.
* No validation on ChrM
* Settings | Add PON to Kit | File browser BSSH integration does not support searching by file name.
* Settings | Add PON to Kit | No pagination, making it difficult to add files without knowing their exact path.
## Fixed Issues:
* End-to-End Cyto Array | Fixed an intermittent issue causing case creation failures for E2E cyto workflows.
* Pipeline | Transcript Prioritization Logic | RNA gene list has been updated.
* Removed genes with weak evidence: HELLPAR, LINC00237, MEG3, LINC00299, GNAS-AS1
* Added genes with new evidence: CHASERR, MIR17HG, RNU2-2 (Limitation as this is not available in VEP:Refseq), RNU5A-1, RNU5B-1, TRU-TCA1-1
* Pipeline | Annotations | Fixed an issue where DGV annotations did not differentiate between DEL and DUP events.
* Variant Page | Related Cases | Fixed component to better support SV insertions, STR and ROH.
* Variant Page | Evidence | ACMG Automation for SNVs | Updated logic so that PM5 and PS1 cannot be applied to the same variant, as per the guidelines.
* Variant Page | Evidence | ACMG Automation for SNVs | Fixed a bug so that intermediate/indeterminate REVEL score *does not support* BP4, as per the guidelines.
* Curate | Fixed a caching issue that prevented some variants from getting annotations when uploaded into Curate.
## Known Issues:
* Add New Case | For Whole Genome cases, the region of interest BED filter does not filter out CNVs from the CNV and CNV-SV callers.
* Add New Case | API | When sending due date please use UTC time, customer time zone is not taken into account with API, only through the UI.
* Add New Case | Replacing a sample in the UI will not change the visible sample name.
* Add New Case, Edit Case | The virtual panel, boosted gene, carrier analysis selector is clickable on the entire row and not just the radio button and text.
* Add New Case | When ingesting cases with DRAGEN SMN caller data, if the CN=Null, case will fail. Fix planned for the October 2025 patch.
* Edit Case | Reanalysis | If HPO terms were updated between analyses, the reanalysis will not automatically map previous HPO terms to new ones.
* Edit Case | Reanalysis | Custom disease is not saved when a case is reanalyzed.
* Cases Page | Illumina Clouds | Users that have been removed from workgroups in IAM can still be added as participants to a case. They will not have access to the software, and there is no security/access risk.
* Cases Page | Reupload fails for JSON files.
* Lab Tab | Insufficient coverage export will not work via UI or API if an included gene does not have a start or end position in NCBI.
* Lab Tab | Incorrect message is displayed when gene has no coverage information. Will be fixed to show ‘No coverage information available for this gene’.
* Lab Tab | When a gene is removed from the knowledge graph, no coverage will be shown, unless gene is removed from gene list.
* Lab Tab | Open in IGV desktop only works if case has been previously linked to IGV desktop from the analysis tools.
* Lab Tab | % BP calculation can be slightly and rarely misleading due to pipeline rounding calculation to two decimal points.
* Lab Tab | Average sample quality bar can be misplaced in the UI, data is correct.
* Analysis Tools | Search | Searching for ‘chromosome: position ref > alt’ is not yet implemented for CNVs.
* Analysis Tools | gnomAD allele frequency rounding inconsistent with Variant Page rounding which is to 4 digits.
* Analysis Tools | Filters | Not all AI modes are available for filtering in Evidence & Tags, advanced mode. Missing Carrier Analysis and Incidental.
* Analysis Tools | Filters | Filtering by User Tags will return AI results.
* Analysis Tools | ‘Last’ button on pagination does not work. Fixed in new analysis tools table.
* Candidates Page | Compound het SNV-CNV variants will not display the automated CNV classification. Workaround – view variants from analysis table.
* Variant Page | Variant Interpretation | Load from Curate | Only a variant is displayed in this component, even if there are several overlapping variants.
* Variant Page, Analysis Tools | When multi-tag is enabled in an account, AI tags appear to be tagged by a user, when a user also tags a variant.
* Variant Page | Summary Tab | gnomAD AF, Max AF and hom/hemi counts for SV INS variants are missing from summary tab but available in Population Statistics section.
* Variant Page | Gene-related disease & Evidence Graph | For CNVs, editing the gene-related disease does not change in evidence graph despite a warning message that it will.
* Variant Page | Clinical Significance | Gene-related disease component shows matching/unmatching disease phenotypes, but can also show patient phenotypes erroneously.
* Variant Page | Clinical Significance | For reanalyzed cases, network classified variants may appear as N/A for cases on GRCh38.
* Variant Page | Quality | Allele distribution chart for reference variants does not work for non-proband case members.
* Variant Page | Visualization | Simple/Advanced selectors will not work for locally uploaded BAM files.
* Variant Page | Visualizations | Curate link isn’t working for Curate track variants.
* Variant Page | Visualizations | Load to desktop IGV | Not working for test subject VCF or ClinVar SV file.
* Variant Page | Population Statistics does not display mtDNA organization DBs even when the case is correctly annotated with the data.
* Variant Page | Connected Variants | Some compound heterozygous connections spanning multiple genes might not be shown in component. They will be captured in filters.
* Variant Page | ACMG Automation | Reclassify is only available for cases that were run with pipeline v37.0, reclassify button might be visible for cases not eligble.
* Variant Page | ACMG Automation | When manually changing a tag status from inactive to active and back again, tag status might be incorrect.
* Variant Page | ACMG Automation | Evidence is missing Emedgene auto-calculation icon when user makes a change.
* Variant Page | ACMG Automation | Evidence is missing user icon when user makes a change.
* Variant Page | ACMG Automation | In the SNV auto classification component, when changing answers to questions, clicking cancel, and then going back to the tag, intermittent caching issues cause the changes to be shown despite the cancel. The data isn’t being saved, this is a UI issue only.
* Report | Download button is not visible in the User Interface in V37+, although the download is still available.
* Curate | Searching for a variant causes the related cases to disappear even when search is removed. Work around is to refresh.
* Curate | Large CNVs do not have a gene-related disease card even if a gene related disease appears in Analyze.
* Curate | ACMG | Does not store tag strength, questions and supporting evidence for cases that were run with pipeline V35 and V36.
* Curate | Orphanet link structure has changed and does not work in Curate (fixed in Analyze).
* Settings | When editing preset filters that contain SpliceAI scores, UI will throw an error message even if filter is saved correctly.
* Organization Settings | API Gene Lists | Does not support NCBI only export/import. This is supported from the UI.
* Organization Settings | Gene Lists | For organizations with thousands of gene lists, UI component might time out occasionally.
* Organization Settings | Tags | Emedgene converts all tags to first letter capitalized, and rest of tag in lowercase. Underscore is converted to whitespace.
* Organization Settings | Setting analysis column order, some columns are missing: AI rank, Variant length, Manual classification, Network classification, Historic AF %, Historic AF #, Noise AF %, Noise AF #.
* Organization Settings | Set mandatory fields - does not work from the UI. Please contact support if you’d like to configure these fields for your account.
* Dashboard | Diagnostic Yield includes Uncertain as Resolved.
* API | sample\_validation\_info does not work in V38 and above.