| Patches | Date |
|---|---|
| V36.10 | February 18, 2026 |
| V36.9 | November 3, 2025 |
| V36.8 | August 11, 2025 |
| V36.7 | June 25, 2025 |
| V36.6 | May 21, 2025 |
| V36.5 | April 6, 2025 |
| V36.4 | March 4, 2025 |
| V36.3 | February 19, 2025 |
| V36.2 | December 11, 2024 |
| V36.1 | October 16, 2024 |
| Type | DRAGEN Output | FASTQ | VCF (BYOD) | Notes |
|---|---|---|---|---|
BAM/ CRAM | In-.BAM/.CRAM Out - .CRAM | ✓ | ✓ | Requires .bai in same folder |
| Small variants | In-vcf/gvcf Out-hard-filtered.gvcf.gz | ✓ | ✓ | Targeted caller variants are removed and ingested via the targeted vcf. |
| SV del/dup/ins | sv.vcf.gz | ✓ | ✓ | VNTR caller outputs are removed from the SV output and not supported on Emedgene yet. |
| CNV | cnv.vcf.gz | - | ✓* | *The new CNV-SV merged file is also supported. Do not use both the CNV and CNV-SV file. |
| CNV-SV | cnv_sv.vcf.gz | ✓ | ✓* | *The new CNV-SV merged file is also supported. Do not use both the CNV and CNV-SV file. |
| STR | repeats.vcf.gz | ✓ | ✓ | Do not use the ExpansionHunter SMN caller, this will fail the case. |
| MRJD | mrjd.hard-filtered.vcf.gz | ✓ | ✓ | |
| Targeted Callers | Targeted.vcf | ✓ | ✓ | GBA, HBA, CYP21A2 w/o CNV, supported. SMN is supported from the targeted JSON. |
| SMN 1/2 | Targeted.json | ✓ | ✓ | Supported from targeted.json starting in 4.3. |
| Ploidy | ploidy_estimation_metrics.csv | ✓ | ✕* | Security requirements prevent the ingestion of csv files at this time, can be pushed in tar. |
| Star Allele | Targeted.json | ✓ | ✓ | Star allele caller, CYP2D6 & CYP2B6 are supported. SMN is also supported. |
| QC metrics | mapping_metrics.csv | ✓ | ✕* | Security requirements prevent the ingestion of csv files at this time, can be pushed in tar. |
| QC metrics | bed_coverage_metrics.csv | ✓ | ✕* | Metrics file containing FASTQC information. |
QC metrics/ TAR | *.metrics.tar.gz | ✕ | ✓* | DRAGEN report for customers starting from VCF. Only available via API. Tar file must contain one of the following. METRICS_PATTERNS = [ r'.csv$', r'.tsv$', r'.counts(.gc-corrected)?(.gz)?$', r'.(ploidy|repeats).vcf(.gz)?$', ] |
| ROH Viz | roh.bed | ✓ | ✓ | |
| BAF BigWig | hard-filtered.baf.bw | ✓ | ✓ | B-Allele frequency (BAF) output. |
| TNS BigWig | tn.bw | ✓ | ✓ | Bigwig representation of the tangent normalized signal. |
| Target Counts BigWig | target.counts.bw | ✓ | ✓ | BigWig representation of the target counts bins. |
| ICLR small variants | combined_iclr_sbs.phased.vcf | ✕ | ✓ | Phased small variant VCF from combined short and long reads. |
| ICLR SV | combined_iclr_sbs.sv.vcf | ✕ | ✓ | Structural variant VCF from combined short and long reads. |
| ICLR BAM | LongRead.haplotyped.BAM | ✕ | ✓ | Aligned Illumina Complete Long Read in BAM format. |
| Gene Name | Condition | Application | Emedgene |
|---|---|---|---|
| PMS2 | Lynch Syndrome | Pharmacogenomics | ✓ |
| SMN1 (small variants) | Spinal Muscular Atrophy | Carrier screening | ✓ |
| STRC | Nonsyndromic hearing loss | Carrier screening | ✓ |
| NEB | Nemaline myopathy | Carrier screening | ✓ |
| TTN | Cardiomyopathy | ACMG secondary, NBS | ✓ |
| IKBKG | Incontinentia pigmenti, Hypohidrotic ectodermal dysplasia | Newborn screening | ✓ |

| Gene Name | Condition | Application | Emedgene |
|---|---|---|---|
| CYP2D6 | NA | Pharmacogenomics | ✓ |
| SMN1 | Spinal Muscular Atrophy | Carrier screening | ✓ |
| GBA | Gaucher’s Disease | Carrier screening | ✓ |
| CYP2B6 | NA | Pharmacogenomics | ✓ |
| HBA1/2 | Alpha-thalassemia | Carrier Screening | ✓ |
| CYP21A2 | Congenital Adrenal Hyperplasia | Carrier Screening | ✓ |
| RHD/RHCE | RH blood type | Blood Typing | ✕ |
| LPA | Cardiac disease risk | Cardiovascular Disease | ✕ |
| Variant Type/Caller | Emedgene Quality Calculations |
|---|---|
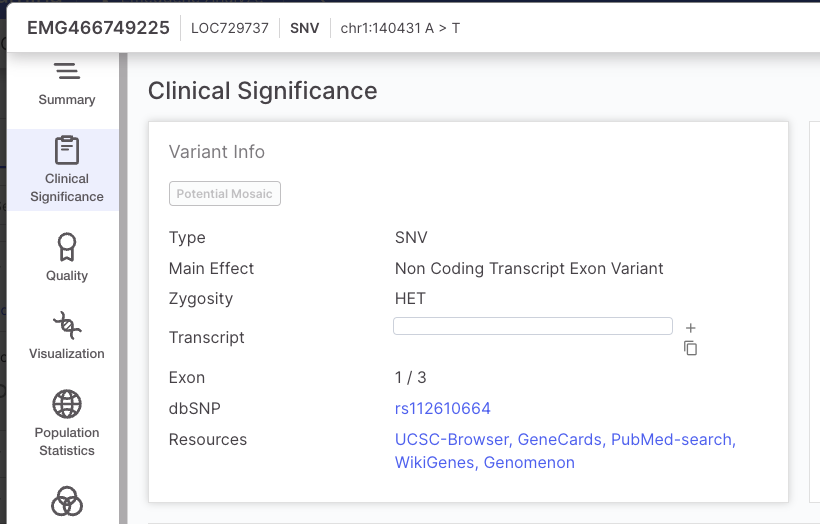
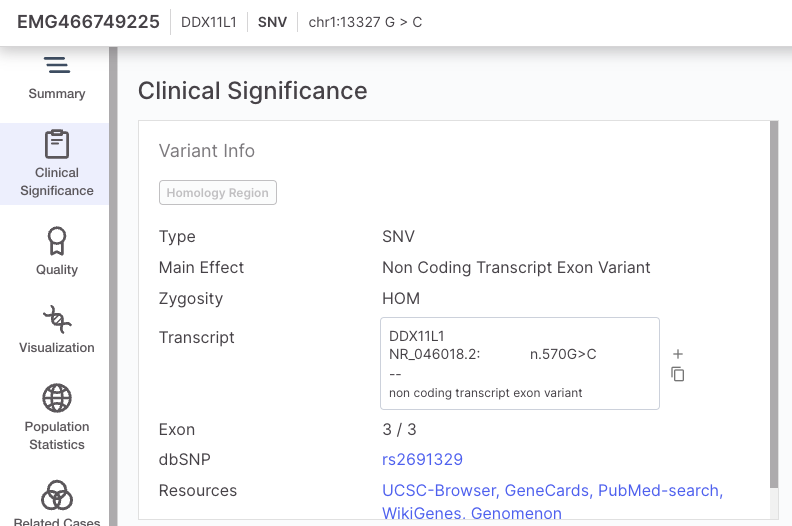
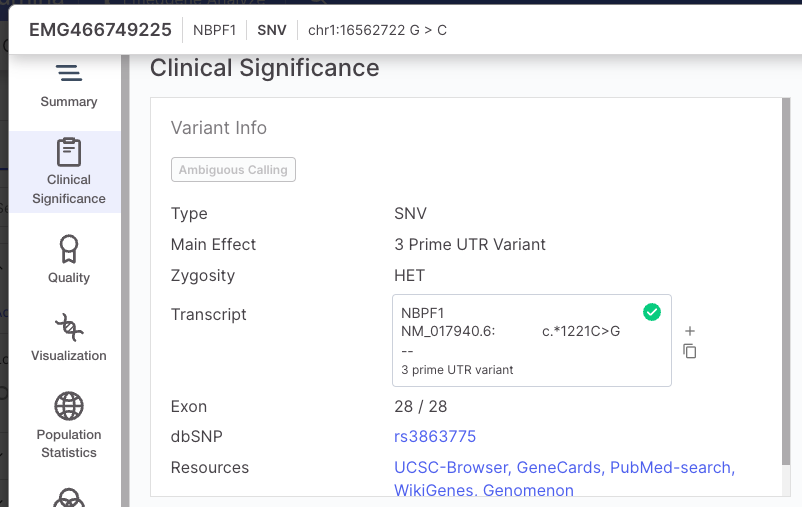
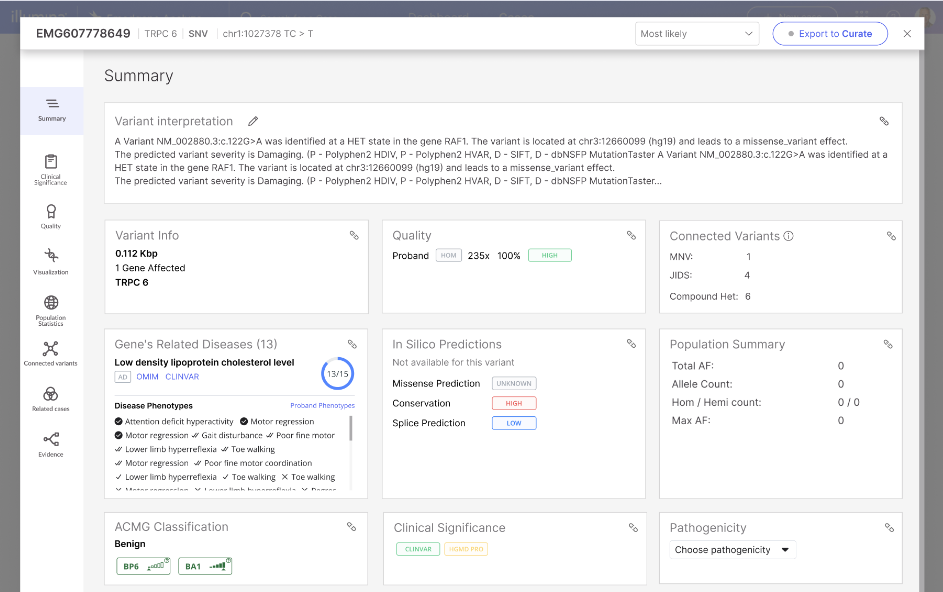
| SNV and small InDels | A variant is designated as Low quality if: · The VCF/FILTER is not "PASS" OR the VCF/QUAL is less than 3 A variant is designated as High quality if: · The VCF/FILTER is "PASS" AND the VCF/QUAL is greater than 30 All other variants are categorized as Moderate quality. The VCF Filter value will be presented in the Variant Page | Quality tab. |
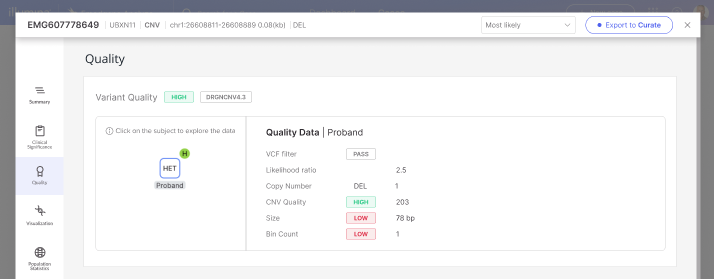
CNVs (called by the CNV_SV caller for genomes) | A variant will be designated as Low quality if: · The VCF/FILTER is not "PASS" AND INFO field SVCLAIM = D A variant will be designated as High quality if: · The VCF/FILTER is "PASS" AND (INFO field SVCLAIM = D OR INFO field SVCLAIM = DJ) AND QUAL > 100. All other variants are categorized as Moderate quality. |
CNVs (called by read-depth caller) | A variant will be designated as Low quality if: · The VCF/FILTER is not "PASS" A variant will be designated as High quality if: · The VCF/FILTER is "PASS" AND VCF/QUAL is greater than 30 All other variants are categorized as Moderate quality. |
| SVs | A variant will be designated as Low quality if: · The SVLEN is greater than 50 kb OR the VCF/FILTER is not "PASS" A variant will be designated as High quality if: · The VCF/FILTER is "PASS" AND VCF/QUAL is greater than 500 All other variants are categorized as Moderate quality. |
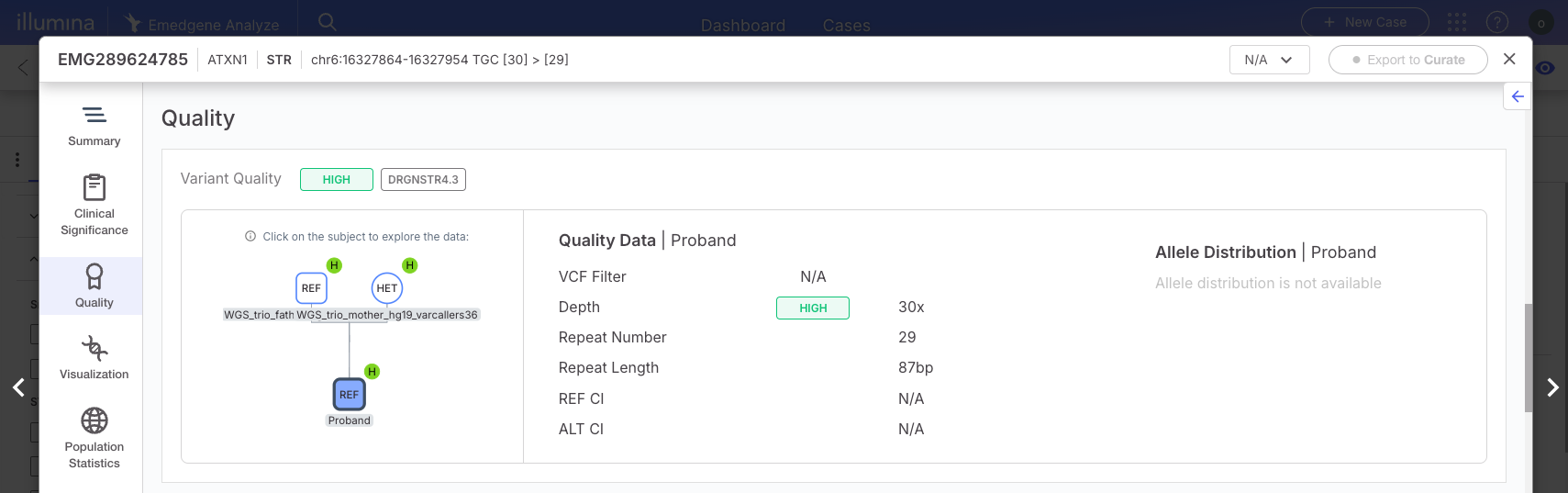
| STR variants | A variant will be designated as Low quality if: · The VCF/FILTER is not "PASS" A variant will be designated as High quality if: · The VCF/FILTER is "PASS" Additional STR loci will always have low quality: ARX, HOXA13 |
| MRJD caller variants | A variant will be designated as Low quality if: · INFO field contains 'JIDS' A variant will be designated as High quality if: · FILTER=PASS and INFO field does not contain 'REGION_AMBIGUOUS' All other variants are categorized as Moderate quality. |
| Targeted caller variants | For variants with no JIDS ID: A variant will be designated as Low quality if: · Filter is not PASS A variant will be designated as High quality if: · FILTER=PASS For variants with a JIDS ID or a Recombinant flag: Always low. |